
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| The Hemoglobinopathies |


|
|
Read More
Date: 10-11-2015
Date: 11-11-2015
Date: 10-11-2015
|
The Hemoglobinopathies
INTRODUCTION
Hemoglobinopathies are classic models for study of molecular diseases as practically all types of mutations are observed in hemoglobin disorders. These are the most common genetic disorders in the world, (approximately 25,000 persons are born each year). They have very high mortality and morbidity thus are of major concern.
STRCTURE OF HEMOGLOBIN
Hemoglobin is found exclusively in red cells where its main function is to transport oxygen from the lungs to the capillaries of tissues. There are three major types of hemoglobin, HbA, HbA2, and HbF. Each of the different types of hemoglobin is a tetramer composed of two a-globin like peptides and two P-globin like peptides. Each globin chain is associated with a heme group (Fig. 1). Heme is the iron-containing compound that combines with oxygen.
1- HbA, Adult hemoglobin (α2β2): HbA forms 90% of the fraction of total hemoglobin, and is the major hemoglobin in adults. It is composed of four polypeptide chains, two a chains and two P chains.
2- HbA2 (α2β2): HbA2 forms 2-5% of the fraction of total hemoglobin and is composed of two a chains and two δchains. It first appears about 12 weeks after birth.
Fig. 1: Structure of hemoglobin
3- HbF fetal hemoglobin,(α2γ2):
HbF forms less than 2% of the fraction of total hemoglobin. It consists of two a chains identical to those found in HbA, and two y chains. The y chains are members of the P-globin gene family. HbF is the major hemoglobin found in the foetus and newborn. During the last months of fetal life, HbF accounts for 60% of the total Hb in the erythrocyte. In the first few weeks after conception, embryonic hemoglobin, (ϭ2Ɛ2, Hb Gower 1) is synthesized by the embryonic yolk sac. Within a couple of weeks the fetal liver begins to synthesize HbF and then the bone marrow takes over. HbA synthesis starts in the first month of pregnancy, and gradually replaces HbF (Fig. 2).

Fig. 2: Hemoglobin synthesis during prenatal and postnatal period
Organization of the Globin Genes
1- α globin gene family: The a gene cluster lies on chromosome 16 (Fig. 3). It contains two genes for the a-globin chain, the o gene that is expressed early in development as a component of embryonic hemoglobin, and a number of globin like genes that are not expressed (pseudogenes). The a chain has 141 amino acids.
2- β-globin gene family: A single gene for the P-globin chain is located on chromosome 11, (Fig. 3) along with four other P-globin like genes. These include the e gene (expressed early in embryonic development), two γ genes, Gγ, and Aγ, that are expressed in fetal hemoglobin HbF, and the δ gene that codes for the globin chain found in the minor HbA2. The β chain has 146 amino acids.

Fig . 3: a- and p-globin gene regions on chromosome 16 and 11
Synthesis and control of hemoglobin expression
In vitro translation studies with reticulocyte mRNA from normal persons have shown that α- and β-globin chains are synthesized roughly equally. However, studies of globin chain synthesis have also demonstrated that β-globin mRNA is slightly more efficient in protein synthesis than a-globin mRNA and that this difference is compensated for in the red blood cell precursors by a relative excess of a-globin mRNA. From this it seems that the most important level of regulation of expression of the globin genes, like other eukaryotic genes, is likely to occur at the level of transcription.
In addition to the promoter sequences in the 5’ flanking regions of the various globin genes, there are sequences 6-20
kb 5’ to the e-globin gene necessary for expression of various β-like globin genes. This region is called the locus control region, lcr, and is involved in the timing and tissue specificity of expression or switching of the β-like globin genes in development.
Hemoglobinopathies
Hemoglobinopathies are defined as a family of disorders caused by production of a structurally abnormal hemoglobin molecule, or by synthesis of insufficient quantities of normal hemoglobin. Examples of conditions that result from production of hemoglobin with an altered amino acid sequence include HbS (sickle cell anemia) and HbC (HbC disease). The thalassemia syndromes result due to decreases production of normal hemoglobin.
More than 300 Hb electrophoretic variants have been described. About 200 of these variants are single amino acid substitutions resulting from point mutations. The types of mutations seen include'
Missense mutations are seen in HbS, HbC, and HbE, nonsense mutations in Hb Constant Spring, deletion mutations in Hb Freiburg, insertion mutations in Hb Grady and fusion polypeptides that result due to unequal cross over events in meiosis in Hb Lepore and Hb Kenya. Though some of the hemoglobin variants are associated with disease, many are harmless and do not interfere with normal function, and are identified only in the course of population surveys of Hb electrophoretic variants.
Any mutation on the inside of the globin subunits in close proximity to the hem pockets or at the interchain contact areas, can produce an unstable Hb molecule which by precipitating in the red blood cell, damages the membrane resulting in hemolysis of the red blood cell. In addition, mutations can also interfere with normal oxygen transport, leading either to an enhanced or reduced oxygen affinity, or to an Hb, which is stable in its reduced form, the so-called methemoglobin. It is not possible to detect all structural variants of Hb by electrophoretic techniques. This is because only about one- third of the possible Hb mutations produce an altered charge in the Hb molecule and thereby can be detectable by electrophoresis.
SICKLE CELL ANEMIA
An autosomal recessive disorder characterized by the substitution of valine for glutamic acid at position 6 in the P globin chain (HbS). This results in a solubility problem in the deoxygenated state, and upon deoxygenation, the affected RBC changes from a biconcave disc to a crescent or sickle shaped cell. Sickle cell anemia is the most common cause of hemolytic anemia in the black population. It is also common among Greeks, Italians, Saudi Arabians, and certain communities in India. There is an association between heterozygote status and protection against malaria.
This is a heterozygous state where both HbA (55-60%) and HbS (35-40%) are present. Those with the trait are usually asymptomatic unless they are subjected to severe hypoxic stress. Abnormalities include failure to concentrate urine (isosthenuria) and painless hematuria secondary to medullary infarcts. Complications include retinal artery occlusion and splenomegaly.
Sickle Cell Anemia
This is the homozygous state, with two HbS alleles. During the first few months of life, high levels of HbF protect the child and the earliest manifestations occur at 4-6 months of age.
The patient may manifest with symmetrical painful swelling of the dorsal surfaces of the hands and feet (hand foot syndrome). This is due to avascular necrosis of the bone marrow of the metacarpal and metatarsal bones. A vasocclusive crisis can involve the chest, abdomen, back and joints. One fourth are preceded by a viral or bacterial infection. Many factors like dehydration, vascular stasis, acidosis or hypoxia can precipitate episodes. Repeated vasocclusive episodes in the spleen lead to infection and fibrosis and the spleen is not palpable after age 5. Due to the spleen being malfunctional, there is increased susceptibility to infection with encapsulated bacteria. Skin may be involved leading to chronic ulcers in the distal lower extremities. All the patients have isosthenuria and renal failure is common. Hepatic infarcts and cholelithiasis also occur. There is aseptic necrosis of the head of the femur. Biconcave (“fishmouth”) vertebrae are pathognomic of sickle cell disease. Osteomyelitis with Staphylococcus or Salmonella is common. Aplastic crises may be precipitated by infection with parvovirus B19.
The diagnosis is made initially by using metabisulfite (an oxygen-consuming agent), which is added to blood. If HbS is present, cells will sickle. The diagnosis is confirmed by hemoglobin electrophoresis.
Thalassemias
Thalassemia are hereditary hemolytic anomies characterized by decreased or complete absence of one or more of the globin subunits of the hemoglobin molecule.
a- α-thalassemia results from reduced α-globin chain synthesis, usually the result of a gene deletion. Normally there are four a chains.
b- β-thalassemia results from reduced β-globin chain synthesis, usually the result of abnormal DNA sequence due to single base substitutions. Normally there are two p chains.
Normally the synthesis of a and β chains are co-ordinated so that each a globin chain has aP globin chain partner. In the thalassemias the synthesis of either the a globin chain or the β globin chain is defective. Each thalassemia can be classified as a disorder in which no globin chains are produced, called as α0 or β0 thalassemia, or in which some chains are synthesized, but at a reduced rate called α+ or β+ thalassemia. The excess unpaired globin chains are a hazard to the RBC because they produce insoluble tetramers that precipitate, causing membrane damage, and susceptibility to destruction within the reticuloendothelial system.
α Thalassemias
These are defects in which the synthesis of the a globin chains is decreased or absent. Because each individual genome contains four copies of the globin gene, two each on each chromosome 16), there are four levels of globin chain deficiency. If one of the four a globin genes is defective, the individual is a silent carrier because no physical manifestations of the disease occur. If two a-globin genes are defective, the individual is designated as a thalassemia trait, or α-thalassemia minor, and the patient has a moderate hypochromic, microcytic anemia. If three α-globin genes are defective, the individual has hemoglobin H disease with mild to moderately severe hemolytic anemia. If all four of the a globin genes are defective, hydrops fetalis and fetal death result, as a-globin chains are required for formation of HbF. The synthesis of unaffected y and then P chains continues, resulting in the accumulation of γ tetramers in the newborn (γ4 or Hb Bart), or β teramers (β4 or HbH). These variants have a high affinity for oxygen, which is not released to the tissues. The result is severe anemia, heart failure, hepatosplenomegaly, generalized edema and death in utero.
Mutational Basis of a Thalassemia
Restriction mapping studies of the a-globin region of chromosome 16 reveal that there are two α-globin structural genes on the short arm of chromosome 16. The various forms of a thalassemia have been shown to be due to deletions of one or more of these structural genes. Deletions of the α-globin genes in a thalassemia are believed to occur as a result of unequal crossover events in meiosis. These events are more likely to occur where genes with homologous sequences are in close proximity. Support for this hypothesis comes from the finding of the other product of such an event, that is persons with three a-globin structural genes located on one chromosome.
β Thalassemias
In the β thalassemias, synthesis of β-globin chains is decreased or absent, whereas P-globin chain synthesis is normal. αglobin chains cannot form soluble tetramers, and therefore precipitate causing the premature death of cells destined to become mature red cells. Because there are only two copies of the globin gene, individuals with β gene defects have either the β thalassemia trait or β thalassemia minor if they have one defective gene or β thalassemia major, if both genes are defective. Because the β-globin gene is not expressed until late fetal gestation, the physical manifestations of β thalassemia appear only after birth.
β Thalassemia trait or β thalassemia minor
The growth and development of patients is normal. There is mild anemia and elevation of HbA2. No treatment is necessary.
β Thalassemia major
Also known as Coolies anemia, or homozygous β thalassemia. Molecular defects range from complete absence of the b globin gene chain synthesis (β0 β0), to partial reduction of the gens product at the affected locus. Beginning in the first year of life, the infant develops a progressively severe hemolytic anemia with hepatosplenomegaly and bone marrow hyperplasia. The bone marrow hyperplasia produces features such as tower skull and frontal bossing. Death occurs due to congestive failure unless the patient is supported by blood transfusions. HbA is markedly decreased and HbF forms30-90% of the total Hb. The treatment includes repeated transfusions and the regular daily use of iron-chelating drugs, such as desferrioxamine.
Mutational basis of β thalassemia
Restriction mapping studies have shown that β thalassemia is rarely due to a deletion, and DNA sequencing has often been necessary to reveal the molecular pathology. A wide variety of different mutations, which include point mutations, insertions and deletions of one or more bases, have been shown to be responsible. These occur at a number of places, both within the coding and the non-coding portions of the β-globin genes as well as in the 5’ flanking promoter region, the 5’ capping sequences and the 3’ polyadenylation sequences (Fig. 4). The various types of mutations causing β thalassemia are often unique to certain population groups and can be considered to fall into five main functional types.
Transcription Mutations
Mutations in the 5’ flanking TATA box or the promoter region of the β-globin gene can result in reduced transcription level of the β-globin mRNA.
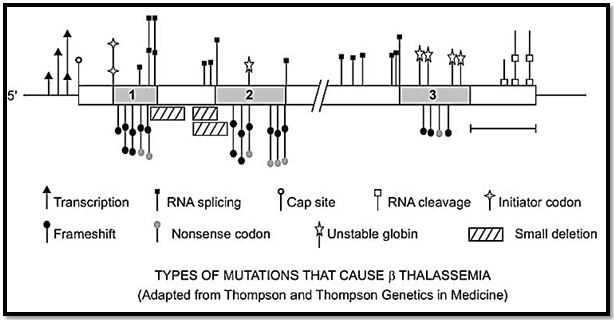
Fig. 4: Mutational types of β thalassemia
mRNA Splicing Mutations
Mutations involving the invariant 5’ GT or 3’ AG dinucleotides of the introns in the P-globin gene or the consensus donor or acceptor sequences result in abnormal splicing with consequent reduced levels of P-globin mRNA. In the commonest P thalassemia mutation in persons from the Mediterranean region, the mutation leads to the creation of a new acceptor AG dinucleotide splice site sequence in the first intron of the P-globin gene creating a so-called cryptic splice site. The cryptic splice site competes with the normal splice site leading to reduced levels of the normal β-globin mRNA. Mutations in the coding regions of the P-globin region can also lead to cryptic splice sites.
RNA Modification Mutations
Mutations in the 5’ and 3’ DNA sequences, involved respectively in the capping and polyadenylation of the mRNA, can result in abnormal processing and transportation of the β-globin mRNA to the cytoplasm with consequent reduced levels of translation.
Chain Termination Mutations
Insertions, deletions and point mutations can all generate a nonsense or chain termination codon, resulting in the premature termination of translation of the β-globin mRNA. This will result in the majority of instances in a shortened β-globin mRNA, which is often unstable and more rapidly degraded with consequent reduced levels of translation of an abnormal P-globin.
Missense Mutations
Missense mutations, which lead to a β-globin chain, which is highly unstable, rarely result in β thalassemia. An example is Hb Indianapolis.
δβ Thalassemia
In δβ thalassemia there is underproduction of both the δ- and β-globin chains. Persons homozygous for δβ thalassemia produce no δ- or β-globin chains. Although one would expect such persons to have a family profound illness, they are only mildly anemic, due to an increased production of γ-globin chains, with Hb F levels being much higher than the mild compensatory increase seen in homozygotes for β thalassemia.
Mutational basis of δ β thalassemia
δβ thalassemia has been shown to be due to extensive deletions in the β-globin region involving the δ- and β-globin structural genes. Some deletions extend to include the Aγ-globin gene so that only the Gγ-globin chain is synthesized.
Hereditary persistence of fetal hemoglobin
Hereditary persistence of fetal Hb, or HPFH, in which there is persistence of the production of fetal Hb into childhood and adult life is included in the thalassemias. Most forms of HPFH are in fact a form of δβ thalassemia in which continued γ-chain synthesis compensates for the lack of production of δ- and β-globin chains. In persons with hereditary persistence of fetal Hb, the fetal Hb accounts for 20-30% of total Hb in heterozygotes and 100% in homozygotes. This is not associated with any symptoms and was originally considered more of a scientific curiosity than a medical problem.
Mutational basis of HPFH
Some forms of HPFH have been shown to be due to deletions of the δ- and β-globin genes. Analysis of the non-deletion forms of HPFH has shown point mutations in the 5’ flanking promoter region of either the Gγ or Aγ globin genes near the CAT box sequence involved in the control of expression of the hemoglobin genes .
References
Purandarey , H. (2009) . Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
للعاملين في الليل.. حيلة صحية تجنبكم خطر هذا النوع من العمل
|
|
|
|
|
|
|
"ناسا" تحتفي برائد الفضاء السوفياتي يوري غاغارين
|
|
|
|
|
|
|
نحو شراكة وطنية متكاملة.. الأمين العام للعتبة الحسينية يبحث مع وكيل وزارة الخارجية آفاق التعاون المؤسسي
|
|
|
















