آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| DEVELOPMENTAL GENETICS |


|
|
Read More
Date: 10-11-2015
Date: 28-10-2015
Date: 12-11-2015
|
DEVELOPMENTAL GENETICS
INTRODUCTION
Progress in in vitro fertilization has helped many couples achieve parenthood. The process of embryonic development is very complex and depends on the genetic and environmental factors at the time of fertilization, which occurs when the egg and sperm meet at the optimal time of a woman’s menstrual cycle. Fertilization takes place in the Fallopian tubes, and the fertilized egg contains the full complement of maternal and paternal genes. With the process of cell division, this fertilized egg forms a small cluster of cells, which are undifferentiated. With appropriate environmental interaction and with an inherent genetic constitution, a cell differentiates, and by the end of 12 weeks from the first day of the last menstrual period (LMP), the foetus is formed. After formation, maturation of the various physiological processes takes place and growth is established. The study of human development from fertilization to the various foetal stages is the field of embryology. The field of developmental genetics involves study of the genetic mechanisms behind this development.
MAIN EVENTS IN THE DEVELOPMENT OF A HUMAN FETUS
There are three main stages in prenatal life, pre-embryonic, embryonic and foetal (Fig. 1).
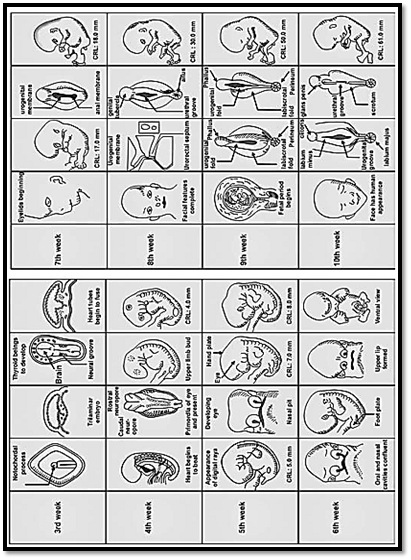
Fig. 1: Important landmarks in fetal development
Fertilisation occurs when the male and female gametes fuse in the Fallopian tube. The number of spermatozoa reaching a single egg is over 100 million. They are deposited in the female genital tract and reach the site of fertilization. Of these only a single spermatozoon is successful in penetrating the corona radiata and zona pellucida of the oocyte. The sperm pierces the oocyte, and it is only then that the second meiotic division takes place. This completes the process of meiosis. The newly formed cell now consists of two nuclei, which are called pronuclei. Each pronucleus contains a haploid set of chromosomes (23). The pronuclei then fuse and a diploid set of chromosome (46) is restored.
The fertilized egg also known as a zygote goes through a series of mitotic divisions. The two-cell stage is reached by 30 hours, four-cell stage by 40 hours, and the 12-cell stage by 163 hours. This last stage is called the morula. Up to this stage, an embryo is in the fallopian tube and during this period any pathology in the tube may result in an ectopic pregnancy. The field of preimplantation diagnosis involves a study of the genetic material from these pre-embryos.
This pre-embryonic stage of development is successfully achieved in vitro. A process of cell division and cavitation forming a blastocyst further develops in an embryo. The blastocyst consists of an inner cell mass called embryoblast, which forms the embryo proper, and an outer cell mass, which forms the trophoblast. The trophoblast gives rise to the placenta and its membranes. The inner cell mass further divides into bilaminar and trilaminar discs. This occurs between the beginning of the second and the end of the third week of development.
The bilaminar embryo is oriented dorsoventrally with the yolk sac below and amniotic cavity above. Epiblast cells migrate through the primitive streak, and gastrulation begins. The notochord formation demarcates the midline. Embryonic regions differentiate and anterior structures like the forebrain and heart, dorsal structures like the neural crest and neural tube, ventral structures like the foregut, lungs and thyroid and posterior structures like the hindgut and allantois are formed. The body form is completely established by 4 to 8 weeks of gestation. The primitive streak appears at the caudal end of the embryo. The three germinal layers ectoderm, endoderm and mesoderm are now formed. The ectoderm develops into skin, teeth, sweat glands and neural tissue. The mesoderm is divided into three parts, paraxial mesoderm forming the skeleton, muscles and dermis, the intermediate mesoderm forms urogenital tissues and lateral mesoderm forms the heart, limbs, and lateral body wall. The endoderm forms the gastrointestinal system, the pharynx, trachea and lungs at the anterior end and the cloaca and urogenital system at the posterior end. Once the body plan and the germ layers are formed, by 4-8 weeks organ systems are formed by cell growth, differentiation and cell migration. The neural tube is formed next and the neural crest cells migrate to form parts of the nervous system - the sympathetic nervous system, sensory ganglia, and pigment cells. The bone and cartilage part of the branchial arches and face are also formed at the same time. Any error at each stage of this minute developmental process can lead to a developmental defect. For example, disorders of nerve cells of neural crest lead to neurofibromatosis type 1.
MOLECULAR ASPECTS OF HUMAN EMBRYONIC DEVELOPMENT
Three developmental biologists and geneticists, who shared the 1995 Nobel prize for physiology/medicine, Lewis, Volhard, and Wieschaus, described how specialized cells are derived from a fertilized egg in a multicellular organism. Their discovery was the finding of pattern-forming genes that control the overall organization of the body. These genes also control development of body segments and the special features of a fruitfly like its legs and wings.
It is well known that most genes produce proteins called transcription factors. Transcription factors control RNA transcription from the DNA template by virtue of a binding process to specific regulatory DNA sequences. These sequences form complexes, which initiate transcription by RNA polymerase.
Developmental Genes
Genes can get switched on and off by transcription factors which in turn, activate or repress gene expression. It is assumed that transcription factors control many different genes in coordinated sequences, which in turn control basic embryologic processes of apoptosis or programmed cell death. It is also presumed, that these processes are mediated by growth factor cell receptors and chemicals, collectively known as morphogens. We are now aware that morphogenesis is the result of intricately regulated pathways of gene expression. For a normal developmental sequence to take place, appropriate genes should be should be expressed at the correct time and in the correct sequence to produce proteins.
A study of human malformation syndromes has shown that various gene families are responsible for isolated malformations or multiple anomaly syndromes.
Segmentation Genes
Segmentation genes have been studied in insect bodies. Insects have many body segments which are repeated and which differentiate into various structures according to their body position. Three main groups of segmentation determining genes are known and are subdivided according to their mutant phenotypes. They are classified as gap mutants that delete groups of adjacent segments, pair rule mutants that delete alternate segments, and segment polarity mutants, which cause portions of each segment to be deleted and duplicated on the wrong side.
Segment polarity genes are responsible for two morphogenes, hedgehog and wingless and are maintained in evolution. Three mammalian hedgehog homologues are known. They are sonic hedgehog, desert hedgehog, and indian hedgehog. Holoprosencephaly, a developmental defect of the ventral neural tube is a lethal condition. In this condition the forebrain is not divided into cerebral hemispheres. Patients with holoprosencephaly have been shown to have loss of function mutations in the sonic hedgehog gene.
Homeobox (HOX) Genes
Mutations in the class of genes known as the homeotic genes are responsible for major structural anomalies determining segment identity in Drosophila or fruitfly. Development of a leg instead of antenna can occur with such a mutation. Homeobox genes are responsible for spatial pattern development and control. In humans four homeobox gene clusters have been identified, Table 1 each inherited in an autosomal dominant fashion. About 30 HOX genes are known. Homeobox specifies a homeodomain of ~60 amino acids (Fig. 2).
Table 1: Chromosomal location of homeobox gene clusters in humans


Fig. 2 : Homeobox genes in Drosophila mouse and human
Syndactyly and polydactyly occur due to a mutation in the HOXD13 genes (Fig. 3). Synpolydactyly is a rare developmental anomaly in which an additional digit originates between the webbed third and fourth digits. The severity increases in homozygotes, with short metacarpals and metatarsals that appear almost like carpal and tarsal bones.
Paired Box (PAX) Genes
Paired box genes were first identified in Drosophila. Paired box encodes a paired domain of ~130 amino acids. PAX genes often have in addition a type of homeodomain known as paired- type homeodomain. These genes encode DNA binding proteins, which are transcription control factors and are of great significance in developmental processes. Nine PAX genes have been identified so far in mice and humans.

Figs 3A and B: (A) Syndactyly (B) Polydactyly
The development of nervous system and vertebral column is dependent on these genes. In humans loss of function mutations of PAX3 lead to Waardenburg’s syndrome type 1 and PAX6 mutations cause aniridia. Waardenburg's syndrome is inherited as an autosomal dominant condition. The clinical features of the syndrome are sensorineural hearing loss, areas of depigmentation and heterochromia of the iris. Aniridia results from gene deletion involving PAX6 locus on chromosome 11p13.
SRY Genes, (HMG BOX) SOX Genes
SRY genes are Y linked genes that play a major role in male sex determination.
The SOX genes have an SRY like HMG box which encodes a domain of ~ 70 amino acids. The SOX genes are transcription regulators. Genes SOX1, SOX2, and SOX3 are expressed during embryogenesis at specific times. About 15 SOX genes have been identified so far.
In humans, loss of function mutations in SOX19 located on chromosome 17 cause campomelic dysplasia. Campomelic dysplasia is a rare disorder characterized by bowing of long bones, sex reversal in XY males and a poor life span. In situ hybridisation in mouse showed the gene to be expressed in the developing embryo in primordial skeletal tissues and on genital ridges.
T-box (TBS) Genes
These genes play an important role in the development and formation of mesoderm and notochord differentiation in mice. The TBX genes encode a transcription factor, which contains activator and depressor genes. Loss of function or mutation leads to mice with short tails and small sacral vertebrae.
The TBX genes are dispersed in the human genome. Approximately 15 genes are known, and the sequence domain is the T-box which encodes a domain of ~ 170 amino acids. The cluster of genes located on chromosome 12 contains the TBX3 and TBX5 genes. Loss of function mutations in TBX5 cause Holt Oram Syndrome, characterized by congenital heart disease (atrial septal defect) and upper limb reduction, which can present as mild hypoplasia of the thumbs due to absence of forearms.
Zinc Finger Genes
The term zinc finger genes refers to genes with a finger like loop formed by a series of four amino acids forming a complex with zinc. Genes containing zinc finger motifs act as transcription factors.
A zinc finger motif containing gene is GL13 on chromosome 7, and is the cause of two known developmental disorders. Large deletions or translocations, which involve GL13, lead to Greigcephalopolysyndactyly. The clinical features include a large head, and hand and foot abnormalities like polydactyly and syndactyly. Frameshift mutations in GL13 lead to Pallister-Hall syndrome. The clinical features of this syndrome are polydactyly, hypothalamic hamartomata and imperforate anus.
Another gene with zinc finger motifs, WT1 is located on chromosome 11. It is responsible for some cases of Wilm’s tumor and Denys-Drash syndrome. In Denys-Drash syndroms the patient has ambiguous genitalia and nephritis leading to renal failure.
Signal transduction genes
These genes are involved in the processes responsible for extracellular growth factors regulating cell growth and differentiation. The pathway is complex and genetically determined, with intermediary steps being involved. Mutations in these genes cause developmental abnormalities and may also be responsible for malignant processes.
The RET Proto-oncogene
This gene is located on chromosome 10q11.2 and encodes a cell surface tyrosine-kinase. Gain of function mutations lead to thyroid cancer, while loss of function mutations has been identified in 50% of familial cases of Hirschsprung’s disease.
In this disease ganglionic cells fail to migrate to the submucosal and myenteric plexuses of the large bowel. Symptoms appear after birth, when the child suffers from intestinal obstruction, and abdominal distention. Multiple endocrine neoplasia (MEN2) characterized by familial clustering of phaeochromocytoma, medullary thyroid carcinoma and parathyroid adenoma and is caused by mutations in the ret oncogene.
Fibroblast Growth Factor Receptors (FGFRs)
This factor plays a principal role in embryogenesis, cell division, migration and differentiation. Nine fibroblast growth factor genes have so far been identified.
Mutations in these genes are seen in two disorders, craniosynostosis syndromes and achondroplasia. An example of a craniosynostosis syndrome is Apert’s syndrome, (Figs 4A and B) characterized by premature fusion of cranial sutures and hand or foot abnormalities. Apert’s syndrome is caused by mutation in FGFR2, in peptides linking the second and third immunoglobulin loops. Mutations in the third immunoglobulin loop cause Crouzon’s syndrome where the limbs are normal, or Pfeiffer’s syndrome where only the thumbs and great toes are abnormal.
A commonly known skeletal dysplasia is achondroplasia, leading to short stature. The limbs have rhizomelic (proximal) shortening, and the head is enlarged with frontal bossing. The patient has normal intelligence and a normal life span. The mutated gene involved is FGFR3. Another mutation in the proximal tyrosine kinase residues of the FGFR3 gene results in skeletal dysplasia of a similar phenotype, except for a normal size and shape of the head. Thanatophoric dysplasia (Fig. 5) is a lethal skeletal dysplasia caused by mutations in the second and third immunoglobulin domains of FGFR3.
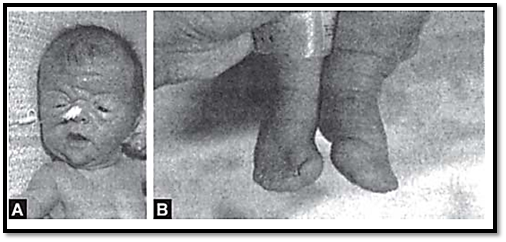
Figs 4A and B: (A) Apert's syndrome (B) Abnormalities in the feet in the same patient
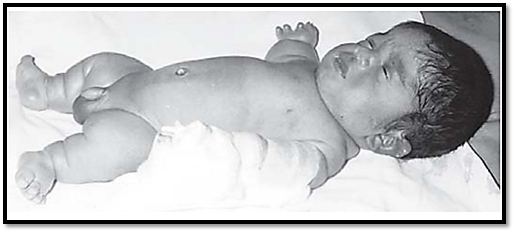
Fig. 5: Thanatophoric dysplasia
Sexual differentiation and x-inactivation
The sex of an individual is determined by the sex chromosomes X and Y. The Y chromosome is responsible for maleness, irrespective of number of X chromosomes. In the absence of the Y chromosome the foetus by default develops into a female.
However, sex determination and differentiation are two different processes and do not occur until 6 weeks of gestation. Up to this stage the gonads possess both the cortex and the medulla but are undifferentiated. The Wolfian and Mullerian ducts are present at this stage and the actual differentiation starts only when the testes determining gene starts an initiation process, which differentiates the so far undifferentiated gonads into testes. The gene responsible for this was discovered in 1990 and is located on the short arm of the Y chromosome adjacent to the pseudoautosomal region. This gene is now labelled as the SRY gene. This SRY (sex regulator gene) gene encodes the code for masculinity (Fig. 6).
The role of the SRY gene in sex differentiation is appreciated by studying individuals with sex chromosomal abnormalities such as the presence of the SRY gene in phenotypic males with a 46 XY karyotype, and the deletion of the SRY gene in XY females. During the process of meiosis 1 all the chromosomes pair with homologous chromosomes that have corresponding gene locations. Sex chromosomes are unequal in size, and have small homologous regions, which can pair at meiosis. However, the SRY gene is in close proximity to the pseudoautosomal region and hence there is a chance that it can get caught in a process of recombination, which is what happens in XX males. The frequency of sex reversal is 1 in 20,000 births. Molecular studies and FISH analysis show the presence of the Y chromosome sequence on the distal end of the short arm of one of the X chromosomes. This region is 140 kilobase pairs long, which is almost .2 percent of the Y chromosome.

Fig. 6 : Generation of XX males and XY females due to recombination events involving the SRY gene
X Chromosome Inactivation
X-linked disorders are expressed in males through carrier females. However, it has been observed that females can have X linked recessive disorders in a mild or a full form (for example female carriers with Duchenne muscular dystrophy). This can occur if there is a structural abnormality of the X chromosome involving that region, or there is involvement of a normal gene in the process of inactivation. The latter is called skewed X inactivation. In females with 46Xr(X) karyotype, typical Turner syndrome features appear as the ring lacks the X sequence, which is normally not inactivated, and appears to be responsible for normal phenotype.
References
Purandarey , H. (2009) . Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
مخاطر خفية لمكون شائع في مشروبات الطاقة والمكملات الغذائية
|
|
|
|
|
|
|
"آبل" تشغّل نظامها الجديد للذكاء الاصطناعي على أجهزتها
|
|
|
|
|
|
|
نقابة تمريض كربلاء تشيد بمستشفى الكفيل وتؤكّد أنّها بيئة تدريبية تمتلك معايير النجاح
|
|
|