
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| CHROMOSOMAL SYNDROMES |


|
|
Read More
Date: 30-10-2015
Date: 11-11-2015
Date: 12-11-2015
|
CHROMOSOMAL SYNDROMES
INTRODUCTION
In this chapter we will discuss selected chromosomal syndromes. These include autosomal trisomy syndromes of chromosome 21 (Down syndrome), 18 (Edward syndrome) and 13 (Patau syndrome) (Table 1) and disorders of the sex chromosomes. In addition we will also discuss the molecular cytogenetics of contiguous gene syndromes. Finally we will discuss chromosomal instability syndromes including ataxia telangiectasia, Bloom syndrome and Fanconi anemia and nucleotide excision repair (NER) syndromes including xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy.
AUTOSOMAL TRISOMIES
Down syndrome (Trisomy 21)
Down syndrome (DS) is the first chromosomal disorder to have been clinically defined and is the most commonly recognized genetic cause of mental retardation.
Clinical Features
The physical characteristics of Down syndrome include upslanting palpebral fissures, loose skin on the nape of the neck, narrow palate, brachycephaly, hyperflexibility, flat nasal bridge, gap between first and second toe (sandal foot deformity), short broad hands, short neck, abnormal teeth, epicanthic folds, short/incurved fifth finger, open mouth and protruding tongue, Brushfield spots of the iris, furrowed tongue and a transverse palmar crease (Fig. 1A).
Table 1: Clinical features of common autosomal trisomies
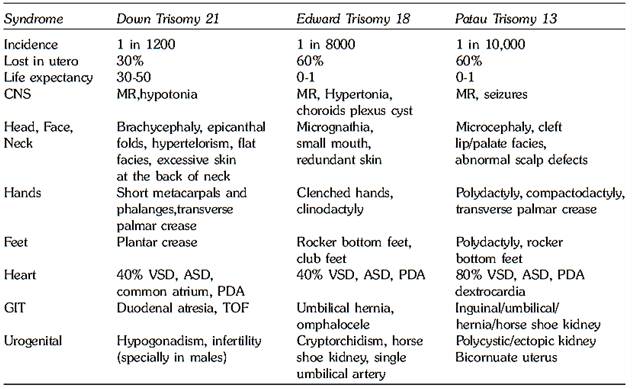

Figs 1A and B: (A) Clinical features of Down syndrome (B) Various chromosomal patterns observed in Down syndrome: trisomy 21,t (14; 21) and t (21; 21)
Mental retardation (IQ varying between 25 and 70) and hypotonia are virtually always present. Congenital heart disease occurs in 40% of individuals, particularly endocardial cushion defects. Heart defects include atrioventricular septal defects, ventricular septal defects and atrial septal defects. Gastrointestinal anomalies like duodenal atresia (double bubble appearance on ultrasound) and Hirschsprung disease are found in 5% of individuals. There is a fifteen to twenty fold increase in the incidence of leukemia in children with Down syndrome, with acute megakaryoblastic leukemia being frequent in cases of acute nonlymphocytic leukemia. Brains of individuals with Down syndrome after the age of 30 show the pathologic, metabolic, and neurochemical changes of Alzheimer disease, and these individuals have a progressive loss in cognitive function.
There is an increased frequency of thyroid dysfunction in newborns and of thyroid autoantibodies throughout life. There is increased susceptibility to infection, due to abnormalities of the immune system, particularly in the maturation and function of T lymphocytes. Males with Down syndrome are invariably infertile, whereas females have decreased fertility but may be capable of reproduction. The principal cause of death in Down syndrome is infection, congenital heart disease and malignancy.
In most cases, trisomy 21 results from an extra chromosome or as part of a Robertsonian translocation or isochromosome. Occasional cases result from a trisomy 21/ diploid mosaicism (Fig. 18.1B). Maternal age plays a role in the incidence of trisomy 21 (Table 2). In 86% of cases, the non-disjunction event occurs in the mother, with the error occurring at meiosis I, 75% of the time.
Prenatal diagnosis by amniocentesis or chorionic villus sampling can detect a fetus with Down syndrome. In addition maternal triple test screening and fetal ultrasound can detect fetuses with Down syndrome.
Table 2: Co-relation between maternal age and Down syndrome

Edward Syndrome (Trisomy 18)
The incidence of this autosomal trisomy in live births is about 1in 8,000. Most fetuses with trisomy 18 are aborted spontaneously. Life expectancy is very low, postnatal survival being very rare. The female to male ratio is seen to be higher, probably due to preferential survival. Primary meiotic nondisjunction is suggested as a cause for this type of trisomy. As with other trisomies maternal age is an important association. The effects are more severe than those of trisomy 21. Features include mental retardation, malformations of the heart, kidney and digestive system, skeletal defects, and failure to thrive. Rocker bottom feet with prominent calcanei and tightly clenched hands with incurved little finger are a characteristic feature (Figs 2A to C). Single palmar crease can be observed along with distinctive dermal patterns on all digits and hypoplastic nails. Either an entire chromosome 18 in addition to the normal complement or a partial trisomy may be observed. Mosaic trisomy may be seen in some cases, which present with milder expression.

Figs.2A to C: Edward syndrome (Trisomy18). (A) Rocker bottom feet and clenched fist. (B) Exomphalos. (C) Metaphase spread showing trisomy 18
Patau’s Syndrome (Trisomy 13)
This is the least frequent of the three common autosomal trisomies with an incidence of 1:10000. The effects are mors severe. Trisomy 13 results from a nondisjunction in meiosis
An unbalanced translocation may be seen in some cases. Severe retardation in physical as well as mental development, along with central nervous system malformations such as arrhinencephaly and holoprosencephaly are associated. The characteristic features are a sloping forehead, ocular hypertelorism, micropthalmia, and coloboma of the iris. The most obvious problems are midline defects such as cleft lip and palate. Figures 3A to C polydactyly and defects in the scalp may be seen. Heart, kidney and digestive tract anomalies are also seen as a common finding. About half the trisomy 13 individuals die within the first month. Survival beyond 3 years is rare.

Figs 3A to C: Patau Syndrome (trisomy13) (A) Microcephaly, microphthalmus, bilateral cleft lip and palate, polydactyly, enlarged kidneys, (B) Scalp defect, (C) Partial karyotype showing trisomy 13
Disorders of sex chromosomes
Cytogenetic abnormalities of the X and Y chromosome occur at a frequency of 1 in 500 live births. Many of the sex chromosomal variations are compatible with life and are among the most commonly seen chromosomal abnormalities. Each has its own set of phenotypic expression, but primarily they involve premature gonadal failure, infertility or abnormal development. The four well-defined syndromes due to numerical abnormalities of the sex chromosomes are are Turner Syndrome (45XO) Klinefelter’s Syndrome (47XXY), 47XXX female and 47XYY male. The structural abnormalities include abnormalities of the X and the Y chromosome and are discussed below after the numerical abnormalities.
Turner Syndrome (45XO)
Turner syndrome (monosomy X) occurs in an estimated 1 to 2% of all clinically recognized pregnancies, although fewer than 1% of these survive to birth. 45,XO is the most common sex chromosomal abnormality.
The typical abnormalities seen in Turner’s syndrome are short stature, gonadal dysgenesis, webbing of the neck, broad chest with widely spaced nipples, and a low posterior hair line (Figs 4A to C). Many a times the condition is not diagnosed until puberty where patients are referred for
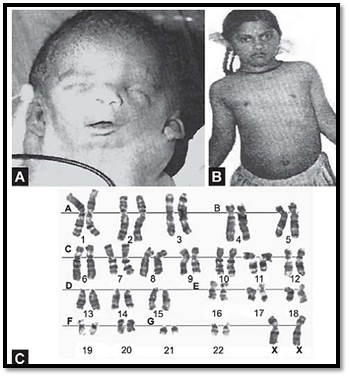
Figs 4A to C: Turner Syndrome (45X) (A) Webbing of neck, (B) webbing of neck, shield shaped chest,cubitas valgus,(C) karyotype showing 45X
primary amenorrhea or shortstature. These patients have a higher frequency of cardiovascular and renal abnormalities. Coarctation of the aorta may be seen in some cases. Postnatal webbing of the neck may be due to cystic hygroma as a result of lymphedema in fetal life. Mental retardation is not a necessary manifestation. Variants of Turner syndrome may be seen, such as those with an isochromosome or a deleted form of chromosome X. They are less commonly seen than monosomy. Cases of this syndrome with a deleted Y have also been observed. Mosaic form of this syndrome along with Klinefelter’s syndrome or with 47,XXX may be seen in some cases. Ultrasound examination particularly of the genital region is suggested in 45,XO chromosomal anomaly (Table 3).
Table 3: Various phenotypes in Turner syndrome variants

Klinefelter’s Syndrome (47XXY)
The incidence of this syndrome is approximately 2:1000 male live births. The main features are increased height with relatively long thin legs. Signs of hypogonadism are seen only when puberty is reached. Gynaecomastia is common, and the penis and testis are smaller than normal. Feminization of the hip may be seen. Patients with this syndrome are usually sterile. Dyslexia is a common manifestation leading to learning difficulties. Variants of Klinefelter’s syndrome are seen, who have more than two X-chromosomes. 48,XXXY or 49,XXXXY (Figs 18.5A and B) patterns may be seen. Though inactive, these additional X chromosomes are usually associated with mental retardation. The phenotype in 49XXXXY is similar to that in Down syndrome. These chromosomal patterns may be observed as mosaics in a normal male or a normal female chromosomal complement. Patients with Klinefelter’s syndrome with azoospermia may benefit by newer technologies in the treatment of infertility like intracytoplasmic sperm transfer technique (ICSI). They need to be counseled and offered prenatal diagnosis, as their progeny will be at risk for a chromosomal disorder.

Figs 6A and B: (A) Partial karyotype from a patient with Klinefelter variant showing XXXY pattern. (B) Buccal smear from the same patient showing 3 Barr bodies
47, XXX Female
They are the female counterparts of Klinefelter’s syndrome seen in males. Rarely tetrasomy X or pentasomy X may be seen. They are phenotypically normal with a taller stature. Patients may develop pubertal changes at an inappropriate age. They are fertile and can bear chromosomally normal children, though there is a greater risk of a meiotic nondisjunction. Patients often have history of repeated fetal wastage. This may lead to the birth of children with other sex chromosomal trisomies. A significant decrease in IQ may be seen. Some have serious learning problems. There may be an effect of late maternal age in some cases. Tetrasomy is associated with serious physical and mental retardation, while pentasomy includes severe developmental retardation and multiple physical defects similar to Down syndrome.
47,XYY Male
This chromosomal constitution is not associated with any observable phenotypic abnormalities. XYY males are very tall and often show behavioral problems such as an excessively violent nature. The intelligence is normal and features are not dysmorphic. The patients are fertile and have nearly no risk of having children with chromosomal abnormalities.
Structural abnormalities of the x chromosome
Deletion Xp Syndromes
Glycerol Kinase Deficiency
This is caused by a deletion of the short arm of the X chromosome at Xp21 (Fig. 7). This is characterized by adrenocortical insufficiency, feeding difficulties and hypogonadotropism. In Glycerol kinase deficiency growth and mental retardation are noted. Patients have elevated urinary glycerol.

Figs 7A to E: Structural anomalies of the X chromosome, (A) XX, (B) i(Xq), (C) deletion Xq, (D) deletion Xp, (E) Translocation t(14;Xq)
Steroid Sulfatase Deficiency/Kallmann Syndrome
The incidence is 1 in 10-15000. It is inherited in an X-linked recessive form. This syndrome is caused by a deletion of chromosome Xp at 22.3. The defects include mutations in the Kal gene, which causes a neuronal migration defect. This is characterized by hypogonadotropic hypogonadism and anosmia. The syndrome may be associated with ichthyosis, sparse hair and conical teeth.
Monosomy Xp
The clinical features are same as that of Turner’s Syndrome. Most patients menstruate spontaneously but menstruation is rarely normal. Patients with del(X)(p21) though menstruate spontaneously half are infertile.
Monosomy Xq
Region of Xq13 or Xq21 is the single most important region for ovarian maintenance. Deletions involving Xq25 27 are less harmful and patient show premature ovarian failure. In this the secondary sexual characters are well developed, the patients have streak ovaries and patient is sterile. Height is not affected in patients with Xq deletion. Isochromosome 46,X,i(Xp) These patients have normal stature and primary amenorrhea.
46,XX Males
These patients usually present as infertility cases. Phenotypically they are males with gynaecomastia and testicular atrophy. The majority of these sex-reversed XX males have inherited a small fragment of the Y chromosome, which includes the SRY gene, transferred to the short arm of one of their X chromosomes.
Structural Abnormalities of Y Chromosome
Structural abnormalities of Y chromosome do not lead to any syndrome but are of great significance in male fertility. The distal portion of Yq shows bright fluorescence with quinacrine stain, and is inherited as a familial marker from father to son. Deletion, isochromosome and elongation of Yp and q, dicentric Y, pericentric Y, ring Y and translocation of Y on an autosome can lead to reproductive loss in a female partner (Figs 8A to C). Relationship between optimal length of Y and reproductive fitness is suggested. Y chromosome micro-deletions occur in 10 to 20% of men with oligoazoospermia. Gene localization is at Yq11 this can be transmitted to male offspring.

Figs 18.8A to C: Structural abnormalities of Y chromosome. (A) Normal Y, (B) del Yq 12, (C) inv (Y)
SEX CHROMOSOMAL ABNORMALITIES: intersex states
These cases are characterized by ambiguous internal and external genitalia. The classification between true and pseudo hermaphroditism is based on the nature of gonads. In true hermaphroditism patients have presence of both male and female gonadal tissue, as mixed gonad (ovo testis) or alternatively an ovary on one side and a testicle on the other side. In pseudohermaphrodism the gonad is either male, (male pseudohermaphrodism) or female (female pseudohermaphrodism). The ambiguity of the genitalia can vary from male to female. Newborns with cryptorchidism or hypospadias are labeled as males. By adulthood, boys may develop gynecomastia or hematuria. Girls present with amenorrhea and hypertrophy of the clitoris. The internal genitalia show persistent Mullerian and Wolfian structures. Cytogenetic studies in true hermaphrodism may show 50% with 46,XX karyotype, 20% with 46,XY karyotype, 20% with XXXY karyotype and remaining 10% with mosaic cell line with one or more additional sex chromosome in one cell line. For example, 47,XXX/46,XX or 49,XXYYY/46,XX.
Male Pseudohermaphrotidism
This is rarely due to chromosomal aberrations. Most of the cases have 46,XY /45,X mosaic cell line. At birth newborns are recognized as male. By puberty axillary and pubic hair develop and a deepening of voice is noted. The built is masculine but genitalia show poor masculinization. A urogenital sinus is always present. The choice of sex rearing must be determined early in life. Male pseudohermaphroditism is also known to occur due to single gene mutation (autosomal recessive or sex linked recessive). The occurrence is usually familial.
Testicular Feminization Syndrome
Earlier known as testicular feminization syndrome, this is not due to a chromosomal rearrangement but due to androgen receptor insensitivity. The phenotype of the individual is female but the karyotype is 46,XY or a mosaic cell line of 46,XY/ 45,X. It occurs due to androgen insensitivity. The molecular defect is in the SRY gene. The inheritance is X-linked recessive and there is a risk to normal female relatives of having an XY female child.
Mixed Gonadal Dysgenesis
This is characterized by a female karyotype with ambiguous external genitalia and hypertrophied clitoris. The gonads and intraabdominal structures are asymmetric. The development of the ducts Mullerian or Wolffian depends on the gonads present on either side.
Female Pseudohermaphrotidism
In a majority of the cases, this occurs due to virilization of a female fetus due to congential adrenal hyperplasia or virilizing hormonal therapy in pregnancy. It is also observed in cases of masculinizing ovarian tumor in the mother.
Contiguous gene syndromes
Contiguous gene syndromes (CGS) are defined as a group of clinically recognizable disorders characterized by a deletion or duplication of a chromosomal segment spanning multiple disease genes, each potentially contributing to the phenotype independently. These genes may be interspersed among other genes whose dosage imbalance has no effect on the phenotype. These syndromes were described before their chromosomal etiology was discovered. Cytogenetic abnormalities are sometimes detectable only by high resolution chromosome analysis or submicroscopic deletions or duplications detectable by molecular methods.
For most autosomal loci, deletion causes a reduction of gene dosage to structural and functional monosomy. Haploinsufficiency for specific genes is implicated for Williams syndrome, Miller-Dieker syndrome and DiGeorge syndrome. Some human genes show exclusive expression from a single parental homologue and no expression from the other homologue, known as genomic imprinting. An example of these is seen in the Prader-Willi and Angelman syndromes. Duplication of chromosomal segments causes increased dosage and gene expression. This is seen in Charcot Maris Tooth disease type 1A and Beckwith-Wiedemann syndrome. Some of the syndromes described above are discussed in more detail below.
Williams’ Syndrome
The chromosomal anomaly in Williams’ syndrome is a deletion of chromosome 7q11.23 including the elastin gene and the LIM kinase gene. The syndrome is characterized by mental retardation, growth deficiency, elfin facies, gregarious personality, infantile hypercalcemia dental and kidney abnormalities, hyperacusis, musculoskeletal and cardiovascular abnormalities. Mutation or deletion of the elastin gene leads to vascular disease. Deletion of LIM kinase gene is thought to account for the impaired visuospatial cognition in William’s syndrome.
Miller-Dieker Syndrome
The chromosomal anomaly in Miller-Dieker syndrome is a deletion of chromosome 17p13.3. It is a multiplemalformation syndroms characterized by lissencephaly and a characteristic facial appearance which which includes a prominent forehead, bitemporal hollowing, a short nose with upturned nares, protuberant upper lip and small jaw (Fig. 9). All affected individuals have profound mental retardation, and about half of them acieve no developmental skills. The LIS1 gene has been identified, and lies within the critical deletion region.

Fig. 9: Miller-Dieker syndrome: Showing dysmorphic features, hypotonia, polydactyly
DiGeorge Syndrome/Velocardiofacial Syndrome
These syndromes are caused by a deletion on chromosome 22 at 22q11.21-q11.23. Symptoms vary greatly between individuals but commonly include a history of recurrent infection, heart defects and characteristic facial features. The patients present with thymic aplasia or hypoplasia, hypocalcemia, hypotonia, developmental delay, sub-mucus cleft palate, dysmorphic facies and cardiac defects of the conotruncal region.
Prader Willi and Angelman Syndromes
These syndromes are due to deletion of 15q11- q13. Prader Willi syndrome occurs due to lack of paternal genes at 15q11 - q13, while Angelman syndrome occurs due to lack of maternal genes at 15q11-q13. Normal banded cytogenetic preparations do not show this deletion but is demonstrated by using fluorescent in situ hybridisation. In Prader Willi syndrome, patients present with hypotonia and polyphagia. The children are short statured and obese (Fig. 10). In Angelman syndrome patients present with inappropriate laughter, convulsions, severe mental retardation, seizures, ataxic gait, and absent speech.
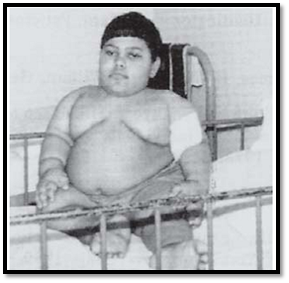
Fig. 10: Prader Willi syndrome; Marked obesity and hypogonadism
Charcot-Marie-Tooth Disease (Type 1A)
The chromosomal anomaly is a duplication of chromosome 17 at 17p12. This region contains the PMP22 gene (peripheral myelin protein gene). This is inherited as a peripheral neuropathy. Patients have weakness, decreased nerve conduction velocities and distal muscle wasting.
Beckwith-Wiedemann Syndrome
The chromosomal anomaly is the duplication of 11p15.5. The syndrome manifests with multiple growth anomalies including hemihypertrophy, macroglossia, exomphalos, visceromegaly, umbilical hernia, gigantism and neonatal hypoglycemia. There is an increased predisposition to several malignancies including Wilm’s tumor, adrenocortical carcinoma, hepatoblastoma and rhabdomyosarcoma.
Some other chromosomal syndromes due to autosomal deletions are described below.
Smith-Magenis Syndrome
The chromosomal anomaly in Smith Magenis syndrome is a deletion of chromosome 17p11.2. Patients have midfacial hypoplasia, brachycephaly, mental retardation, short broad hands, and self-abusive behavior.
Cri du chat Syndrome
The chromosomal anomaly in Cri du chat syndrome is a deletion of chromosome 5p15.2-p15.3. It is characterized in a newborn by a shrill cry resembling the mewing of a kitten. The disorder occurs 1 in 50,000 live births. Clinically patients have hypotonia, microcephaly, moon shaped face, hypertelorism, and micrognathia. Cerebral anomalies, congenital heart defects and renal malformations are sometimes associated. Most cases occur de novo but a chromosomal rearrangement in parents should be investigated (Figs 11A and B).

Figs 11A and B: (A) Cri du chat syndrome, (B) Pedigree showing partial karyotype from the child with 5p-, normal mother (46XX), father with 46XY t(5p-;9q+). CVS from the mother showed the fetus had the same karyotype as the carrier father
Wolf-Hirschorn Syndrome
The chromosomal anomaly in Wolf-Hirschorn syndrome is a deletion of chromosome 4p16.3. Occurrence is relatively rare and is usually de novo, though familial translocations have been seen. Clinically cleft lip and palate, microcephaly, small chin and mental retardation are noted.
Chromosome instability syndromes
These are rare inborn disorders inherited in an autosomal recessive fashion. They have hallmarks characteristic for repair defects or inadequate response to DNA damage. The chromosomes in these conditions show morphological abnormalities in culture. The best-studied syndromes are ataxia-telangiectasia (AT), Bloom syndrome (BS) and Fanconi Anemia (FA). All three disorders display different manifestations of cancer proneness and increased susceptibility to specific mutagens.
Ataxia Telangiectasia (AT)
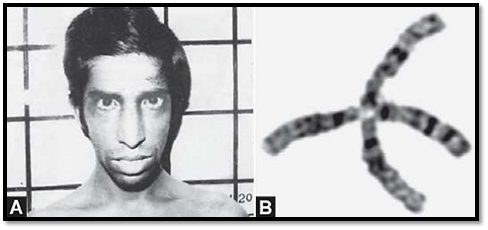
This occurs in 1:40 000 live births in the United States. The carrier frequency is estimated to be 1%. The major clinical features are progressive cerebellar ataxia and telangiectasias of the skin and conjunctiva. There is predisposition to lymphoid malignancies. It is a progressive degenerative disease characterized by cerebellar degeneration, immunodeficiency, and radiosensitivity. Serum alfa- fetoprotein is elevated in 95% of patients. Chromosomal breakage is a feature. Karyotyping reveals characteristic translocations involving chromosomes 14 and 7.
The ATM gene maps to chromsome 11q22-q23. The ATM gene belongs to a large molecular weight family of protein kinases, and the ATM gene product senses double-stranded DNA breaks. When the ATM protein is defective, the signal to arrest the cell cycle is not given, and DNA damage does not get appropriately repaired.
Bloom Syndrome (BS)
Bloom syndrome is an autosomal recessive disorder characterized by proportionate growth deficiency, sun- sensitive, telangiectatic hypo- and hyper-pigmented skin, predisposition to malignancy and chromosomal instability. Patients also have a characteristic facies and head configuration and immunodeficiency, often associated with otitis media and pneumonia. The three major complications are chronic lung disease, diabetes mellitus and cancer. The diagnosis of BS is based on clinical observation. Laboratory confirmation is by cytogenetic demonstration of increased frequency of sister chromatid exchange (SCE) (Fig. 12). The gene for BS has been cloned, and is called the BLM gene on 15q26.1. The protein shows motifs characteristic of DNA and RNA helicases, specifically the RecQ subfamily of DNA helicases.
Figs 12A and B: (A Blooms syndrome, (B) Quadrilateral chromosome observed in Bloom syndrome
Fanconi Anemia (FA)
Fanconi anemia is an autosomal recessive disorder with a predisposition to bone marrow failure and malignancy, particularly acute myelogenous leukemia (AML). FA patients exhibit extreme heterogeneity and may have abnormalities in any orgen system. FA is found in all races and has a carrier frequency of 1 in 300. The diagnostic criterion used is the hypersensitivity of FA cells to the chromosome breaking effect of cross-linking agents such as diepoxybutane (DEB). The hypersensitivity of FA cells to cross-linking agents has been used to assess complementation groups. Complementation groups usually are considered to represent distinct disease genes and for FA four groups represent disease genes namely groups A, C, D and G. The FAC gene (formerly FANCC) maps to 9q22, the FAA gene (formerly FANCA) to 16q24, the FAD gene (formerly FANCD) to 3p22 and the FAG gene (formerly FANCG) gene maps to 9p13. The only cure for the bone marrow failure in FA is transplantation from hematopoetic stem cells from bone marrow or umbilical cord blood.
Nucleotide Excision Repair Syndromes
Three autosomal recessive syndromes are associated with nucleotide excision repair (NER) defect: Xeroderma pigmentosum (XP), Cockayne syndrome (CS), and the photosensitive form of trichothiodystrophy (TTD). In all three conditions, patients have extreme sensitivity to sunlight.
Xeroderma Pigmentosum (XP)
Affected patients (homozygotes) have sun sensitivity resulting in progressive degenerative changes of sun exposed portions of the skin often leading to neoplasia.
All seven NER genes involved in XP (namedXP-A through X-PG) have been cloned and their defect analyzed in patients.
Cockayne’s Syndrome (CS)
Patients with Cockayne’s syndrome have a combination of sun sensitivity, short stature, severe neurologic abnormalities due to dysmyelination, cataracts, dental caries, and a bird like facies. Patients do not display a cancer predisposition. After exposure to UV radiation, patients can no longer perform a certain type of DNA repair called transcription coupled repair. Cockayne’s syndrome spans a spectrum that includes CS type I, the classical form, CS type II also known as cerebro- oculofacial syndrome, CS type III a milder form and the above which includes DNA repair defects. Two genes responsible for Cockayne syndrome have been identified, ERCC6 (10q11) CKN1 on chromosome 5. Mutations in the excision repair cross complementing group 6 (ERCC6) cause CSB which accounts for 75% of cases and mutations in the CKN1 gens cause CSA which accounts for 25% of cases. Both genes code for for proteins that interact with components of the transcriptional machinery and the DNA repair proteins.
Trichothiodystrophy: Affected individuals are short, microcephalic, have sparse, short, thin and brittle hair; a receding chin and small nose giving a peculiar facial appearance. Mental impairment is non-progressive. The hallmark of TTD is sulfur defcient brittle hair and nails, and icthyosis. About half of TTD patients are hypersensitive to UV light, and they have a nucleotide excision repair defect. There are no indications for an increased risk of cancer. Three of the NER genes involved in TTD are XP-B, XP-D and TTD-A.
References
Purandarey, H. (2009). Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
دراسة يابانية لتقليل مخاطر أمراض المواليد منخفضي الوزن
|
|
|
|
|
|
|
اكتشاف أكبر مرجان في العالم قبالة سواحل جزر سليمان
|
|
|
|
|
|
|
المجمع العلمي ينظّم ندوة حوارية حول مفهوم العولمة الرقمية في بابل
|
|
|
















