آخر المواضيع المضافة

 علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية
علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية | Wagner-Meerwein Rearrangements |


|
|
Read More
Date: 10-7-2019
Date: 19-7-2019
Date: 11-10-2020
|
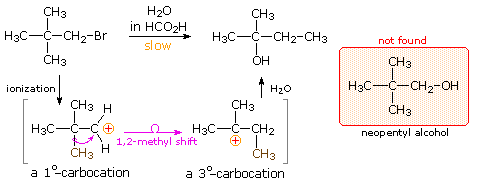
The chemical behavior of neopentyl bromide, 2,2-dimethyl-1-bromopropane, is an instructive place to begin this discussion. The very low SN2 reactivity of this 1º-bromide was noted earlier, and explained by steric hindrance to the required 180º alignment of reacting orbitals. Under conditions that favor SN1 reactivity, such as solution in wet formic acid, neopentyl bromide reacts at roughly the same rate as ethyl bromide. Both of these compounds are 1º-alkyl halides, and for an SN1 reaction the rate determining step requires ionization to a 1º-carbocation. As noted in the carbocation stability order shown below, such carbocations are relatively unstable and are formed slowly. The product from ethyl bromide is ethanol, the simple and direct substitution product, but neopentyl bromide yields 2-methyl-2-butanol instead of the expected neopentyl alcohol. A change in the way the five carbon atoms in this product are bonded to each other has clearly taken place.
|
Once formed, the ethyl cation can only be transformed by a substitution or elimination process. In the case of the neopentyl cation, however, the initially formed 1º-carbocation may be converted to a more stable 3º-carbocation by the 1,2-shift of an adjacent methyl group with its bonding electrons. A mechanism demonstrating such a rearrangement is shown below, and it explains the overall structural changes very nicely.
Increasing the stability of carbocation intermediates is not the only factor that leads to molecular rearrangement. If angle strain , torsional strain or steric crowding in the reactant structure may is relieved by an alkyl or aryl shift to a carbocation site, such a rearrangement is commonly observed. The following examples illustrate rearrangements induced by the strain in a small ring. Although a 3º-carbocation is initially formed, the angle and torsional strain of the four-membered ring is reduced by a methylene group shift resulting in ring expansion to a 2º-carbocation.
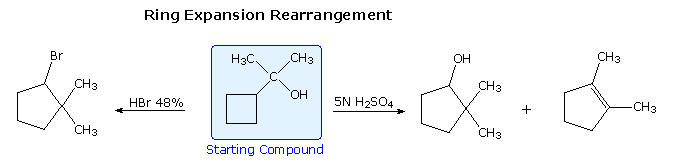
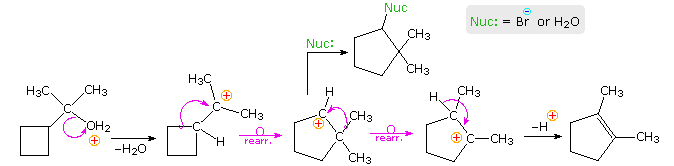
Following the ring expansion step other reactions may take place, depending on the conditions. In aqueous acid the rearranged 2º-carbocation may bond to a water nucleophile, producing a 2º-alcohol, lose a proton to water, giving 3,3-dimethylcyclopentene (not shown), or undergo a second rearrangement to a 3º-carbocation, which then forms 1,2-dimethylcyclopentene. Indeed, it is not uncommon to encounter sequences of rearrangements in more complex compounds, and these may produce products with structures remarkably different from that of the starting compound. The following equation shows one such reaction. A curved arrow representation of the five sequential ring expansion steps will be added to the equation by clicking on the diagram.
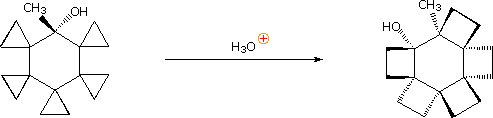
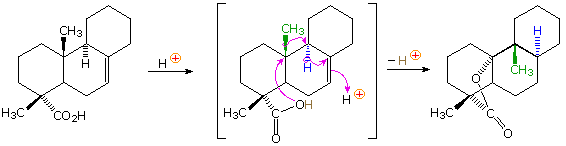
In the terminology of pericyclic reactions, 1,2-alkyl shifts of this kind are classified as [1,2]-sigmatropic shifts. Since this is a two-electron process (the 2 electrons in the relocated sigma bond), the rearrangement is predicted to be suprafacial. Considerable evidence supporting this conclusion has been obtained, as the following example shows. Protonation of the double bond gives a 3º-carbocation. An adjacent hydrogen atom (colored blue) shifts as a hydride moiety to create a new 3º-carbocation, which in turn induces the shift of a methyl group (colored green) with formation of yet another 3º-carbocation. This electrophilic center then bonds to the nucleophilic oxygen of the carboxylic acid function, releasing a catalytic proton to continue the process. Because of the fused polycyclic structure of this compound, the relative orientation of the migrating groups is easily determined, and is seen to be suprafacial. Rearrangements consisting of consecutive 1,2-shifts often take place in a concerted, and therefore stereospecific fashion; however, it must not be assumed that the group shifts are simultaneous. Each shift involves a separate transition state in which the positive charge is delocalized over the migration terminus, origin and migrating group.
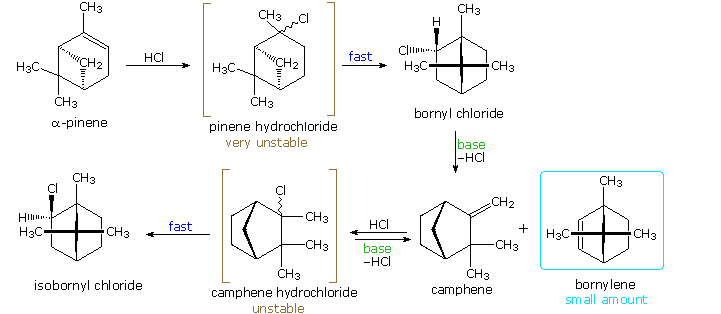
Many of the most interesting rearrangements of this kind were discovered during structural studies of naturally occurring compounds. Among these the terpenes presented numerous remarkable reactions, and the names of two chemists who were instrumental in unraveling their complex transformations, H. Meerwein and G. Wagner, are permanently associated with these rearrangements. The addition of gaseous HCl to α-pinene proved particularly puzzling to these early chemists. Under ordinary conditions, this liquid component of turpentine gave a crystalline C10H17Cl compound, originally called "artificial camphor", now known as bornyl chloride. An unstable isomer, pinene hydrochloride, can be isolated under mild conditions, but it rapidly isomerizes to bornyl chloride. Treatment of bornyl chloride with base gave a crystalline isomer of pinene called camphene, together with small amounts of another unsaturated hydrocarbon (bornylene). Addition of HCl to camphene, in a similar fashion, initially produces an unstable chloride (camphene hydrochloride) which quickly isomerizes to isobornyl chloride, a stereoisomer of bornyl chloride. We now know that bornyl chloride and isobornyl chloride are endo / exo-2-chloro isomers of the 1,7,7-trimethylbicyclo[2.2.1]bicycloheptane system. Structural formulas for these compounds are drawn below, along with camphene, the rearranged elimination product.
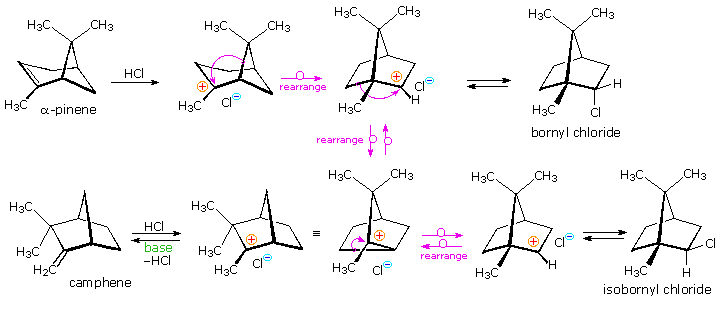
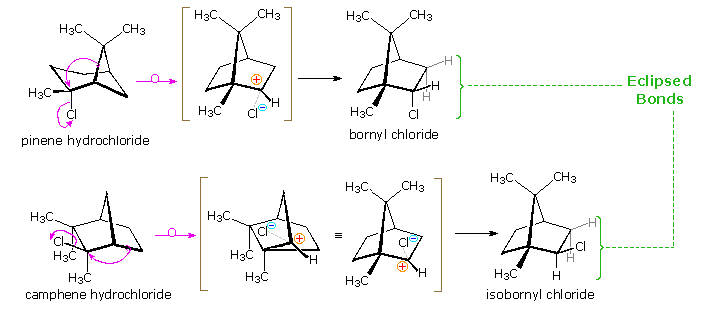
Mechanisms for these rearrangements will be pictured as above diagram. In the new display we see that both pinene and camphene form 3º-carbocations when the double bond is protonated. Rearrangement to a 2º-carbocation is favored by relief of small-ring strain in the case of pinene, and relief of steric congestion in the case of camphene. However, this is an oversimplification which ignores the fact that these reactions take place in nonpolar solvents, and are unlikely to involve discrete, unassociated carbocations. Some of the stereoelectronic effects that influence these reactions will be shown by clicking on the above diagram a second time. Structures for the initially formed unstable hydrochlorides of pinene and camphene are drawn on the left. Optimal orbital overlap of breaking and forming bonds requires rear-side approach of the shifting alkyl group to the site of the leaving chloride anion, in a manner similar to a SN2 reaction. The chloride anion is located on one side of the carbocation formed by the alkyl shift, and immediately bonds to that face of the tricoordinate carbon. In this view of these rearrangements, the chloride anion never escapes the attractive influence of its cationic partner, and the product stereoselectivity is understandable. Lewis acid catalysts (e.g. FeCl3) catalyze these rearrangements, and the product favored at equilibrium is bornyl chloride.
The rearrangement that occurs under base catalyzed elimination conditions reflects the eclipsed configuration of the two-carbon bridge bearing the chlorine atom. Because of this configuration, the anti-coplanar structure favored by the E2 transition state cannot be achieved. Syn elimination gives a small amount of bornylene, but rearrangement to a camphene precursor predominates. Repeated clicking on the above diagram will cycle the displays.
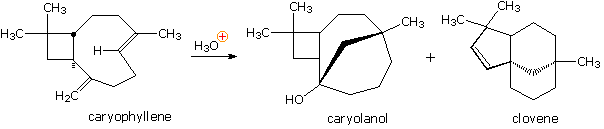
Yet another example of the remarkable acid-catalyzed rearrangements found to occur with terpenes was observed in a study of the sesquiterpene caryophyllene (from oil of cloves). Here it is evident that reactive sites may interact and form bonds from one side of a medium-sized ring to another side. The mechanisms for many such rearrangements have been, and still are studied with great interest.


|
|
|
|
دخلت غرفة فنسيت ماذا تريد من داخلها.. خبير يفسر الحالة
|
|
|
|
|
|
|
ثورة طبية.. ابتكار أصغر جهاز لتنظيم ضربات القلب في العالم
|
|
|
|
|
|
|
سماحة السيد الصافي يؤكد ضرورة تعريف المجتمعات بأهمية مبادئ أهل البيت (عليهم السلام) في إيجاد حلول للمشاكل الاجتماعية
|
|
|