
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Amyloid Precursor Protein |


|
|
Read More
Date: 2-4-2021
Date: 15-12-2015
Date: 10-12-2015
|
Amyloid Precursor Protein
At the molecular level, the necessary, and probably sufficient, cause for the brain dysfunction in Alzheimer's disease is the deposition of aggregated forms of the amyloid b-protein (Ab or bA4), a proteolytic fragment of the amyloid precursor protein (APP), in the brain parenchyma and vascular system (1). In the Western world, dementia is the most common neurological diagnosis and is the third leading cause of natural death, and Alzheimer's disease is by far the dominant cause of dementia (2). It has been estimated that there are 3 million patients in the United States alone with Alzheimer's disease (3). In addition, older patients with Down's syndrome (trisomy 21) tend to develop a neuropathology that is very similar to that seen in Alzheimer's patients (4) The disease is chronic, progressive, and at present untreatable. The progression of the disease begins with loss of short-term memory and disorientation, followed by complete impairment of memory, judgment, and reasoning. Firm diagnosis of the disease can be given only after postmortem examination of the brain, which is characterized by extensive loss of neurons and particular microscopic lesions, which include what are now called “neurofibrillar tangles” and “senile plaques.” The neurofibrillar tangles are composed of paired helical filaments consisting of phosphorylated tau protein, which is normally associated with microtubules. The senile plaques are composed mainly of Ab deposited in the brain parenchyma. Also present in the brain is congophilic amyloid angiopathy from the accumulation of the Ab peptide in the walls of blood vessels. The Ab protein in the senile plaques and in the vascular deposits is present in the form of amyloid (5, 6), long, stable fibrils resulting from the accumulation of precursor peptides that adopt a continuous beta-sheet structure. In this regard, Alzheimer's disease is related to Creutzfeldt–Jakob disease (CJD) and other spongiform encephalopathies, which are also characterized by the presence of amyloid deposits, in that case composed of the prion protein. All known risk factors for Alzheimer's disease appear to influence one or more of the following: (1) the concentration of Ab (7); (2) the amount of the longer, more amyloidogenic Ab chains (8, 9); or (3) the initiation of amyloid formation (10). Increases in any of these factors appear to increase the risk of Alzheimer's disease. Study of the origins of the b-amyloid protein from APP, its aggregation into amyloid fibrils, and the identification of genes involved in its inherited susceptibility are therefore central to understanding of, and devising therapies against, the scourge of Alzheimer's disease.
The amyloid precursor protein (APP), is a 110–130-kDa protein with the features of a transmembrane cell-surface glycoprotein. It is encoded by a gene localized on chromosome 21 encoded by 18 exons. A family of eight transmembrane glycoproteins is generated by alternative splicing of some of these exons (11). The amino acid sequence predicts that the single transmembrane domain of APP is near the C-terminus (Fig. 1) (12). The Ab sequence consists of a maximum 43-residue segment straddling the transmembrane and extracellular portions of the APP chain, and encoded by parts of exons 16 and 17. APP is processed in two distinct pathways : a major nonamyloidogenic route involving proteolytic cleavage within the region carrying the Ab segment to separate the extracellular and membrane-bound domains (13): and an amyloidogenic route, shown in Figure 1, leading to the release of the Ab segment (7). The cleaving of the Ab sequence from an internal site in the APP chain implies that two distinct proteolytic events are required to generate the N-terminus and the C-terminus of the Ab peptide. The proteolytic enzymes involved in processing APP are termed secretases: a-secretase for cleavage of APP within the Ab segment, and b- and g-secretases for cleavage of APP at the N- and C-terminal sides of the Ab segment, respectively. Soluble, C-truncated forms of APP are generated by two pathways: (1) by a-secretases cutting within the Ab sequence, thereby precluding the release of Ab (13); and (2) by b-secretases cleaving near the N-terminus of Ab producing C-terminal fragments containing the complete Ab (14). The generation of intact Ab chains suggests that g-secretases generate the C-terminus of b-amyloid from these C-terminal fragments of APP after release of the transmembrane domain from the lipid bilayer (7). The identity and location of the secretases have proved elusive, but clearly they are of prime importance in Alzheimer's disease, and targets for therapies.
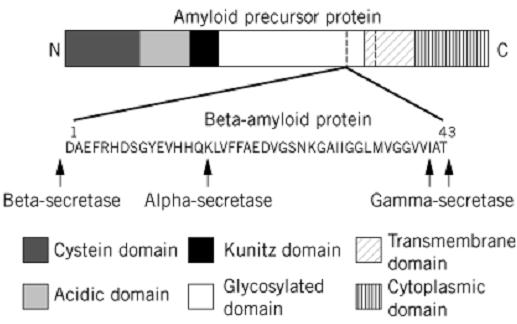
Figure 1. Schematic diagram of the derivation of the 4-kDa b-amyloid protein (Ab) from the 110–130-kDa amyloid precursor protein (APP) by the action of the a-, b-, and g-secretase enzymes. The probable domain structure of APP has been simplified.
Electron microscope and X-ray fiber diffraction analyses, often using synthetic analogs or fragments of the Ab peptide, have revealed the molecular characteristics of the Ab amyloid fibrils. In the electron microscope, different forms of Ab amyloids are seen at different pH values, but the physiological form appears to be represented by a fibril about 90 Å (9 nm) diameter, composed of five or six parallel protofilaments 25–30 Å (2.5–3 nm) in diameter arranged around a hollow core (15). The X-ray fiber diffraction patterns of Ab amyloid (16, 17) show the usual 4.7 Å (0.47 nm( meridional reflection and 10 Å (1 nm) equatorial reflection that demonstrate that the molecular structure of amyloid consists of paired b-sheets running parallel to the fiber axis, with their constituent b-strands perpendicular to the fiber axis. This structure is very similar to that observed in other amyloids produced from a variety of proteins and peptides (see Amyloid). Some shorter Ab chains, such as residues 11–28, particularly when aligned in a strong magnetic field, form pseudocrystals (18), which give more detailed X-ray diffraction patterns that may be capable of yielding greater structural detail and hence begin to define the features that dispose them to form amyloid.
The discovery that Ab is produced and secreted by cells continuously under normal metabolic conditions, and is present in a soluble form in biological fluids (14, 19), suggests that other factors are involved in Ab amyloidosis. It has been proposed that the isolated Ab protein exists in two forms: a soluble form that may represent a normal host protein and a modified amyloidogenic form (20) . These two forms have identical sequences and hence are likely to represent conformation isomers. The soluble form is easily degraded and appears to have an alpha-helical/random coil structure: NMR analysis of synthetic Ab 1–40 (21) has shown that residues 15–23 and 31–35 form a-helices, while the rest of the peptide is in a random-coil conformation, and no stable tertiary structure is present. The amyloidogenic form is more resistant to degradation, has a high b-sheet content, and forms the fibrillar aggregates found in brains with Alzheimer's disease. This behavior is very reminiscent of the normal and scrapie forms of the prion protein, which is also an agent for amyloid brain diseases. The existence of two structural forms for the Ab protein implies the possibility that other proteins may be involved in regulating their interconversion. It is known, for example, that in the rare early-onset (30–60 years of age) form of Alzheimer's disease, the disease has been linked to mutations on chromosomes 1 and 14, in addition to chromosome 21, which carries the APP gene. The genes on chromosomes 14 and 1 have been identified with presenilin 1 (22) and presenilin 2 (23, 24), respectively. The two presenilins have 65% amino acid sequence identity, and hydrophobicity plots predict seven transmembrane regions resembling a structural membrane protein. The genetic variants of presenilin 1 allow the secretion of Ab peptides of longer length, and hence more amyloidogenic (25), possibly through a mechanism involving protein sorting and trafficking of APP.
The more common sporadic and familial late onset (>65 years) form of the disease has been linked with mutations on chromosome 19, subsequently narrowed to the 4 allele of the apolipoprotein E ) apoE) gene (26, 27). These studies have shown that carriers of the 4 allele have an increased risk of developing Alzheimer's disease and that the inheritance of the e4 allele correlates with an increased deposition of Ab amyloid in blood vessels and plaques. The apoE4 isoform differs from apoE3, the most common isoform, by an Arg/Cys substitution at position 112. The apoE protein is known to be one of the proteins associated with amyloid deposits (28). It has been found that apoE can form complexes with synthetic Ab analogues and to enhance amyloid fibril formation by Ab in vitro (29), possibly by a direct physical interaction. Frangione and his colleagues (30) have made the intriguing proposal that apoE can itself possibly form an amyloid-like structure (there is experimental evidence for this in its C-terminal domain), which may be able to induce other proteins such as Ab to misfold into a b-sheet structure that could allow them to be incorporated into a growing amyloid fibril. They term this process “conformational mimicry,” and certainly the most recent analyses of in vivo amyloid formation characterize it as a disease of protein misfolding, which is possibly cooperative.
References
1. A. I. Bush, K. Beyreuther, and C. L. Masters (1992) Pharmacol. Ther. 56, 97–117.
2. A. Ott, M. M. B. Bretaler, and F. van Harskamp (1995) Br. Med. J. 310, 970–973.
3. R. E. Scully et al. (1993) New Engl. J. Med. 324, 1255–1263.
4. K. E. Wisniewski, A. J. Dalton, D. R. Crapper-McLachlan, G. Y. Wen, and H. M. Wisniewski (1985) Neurology 35, 957–961.
5. G. G. Glenner and C. W. Wong (1984) Biochem. Biophys. Res. Comm. 120, 885–890.
6. C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald, and K. Beyreuther (1985) Proc. Natl. Acad. Sci. USA 82, 4245–4249.
7. C. Haas and D. J. Selkoe (1993) Cell 75, 1039–1042.
8. C. Hilbich, B. Kisters-Woike, J. Reed, C. L. Mastrers, and K. Beyreuther (1992) J. Mol. Biol228,. 460–473
9. N. Suzuki et al. (1994) Science 264, 1336–1340.
10. B. Hymen et al. (1995) Proc. Natl. Acad. Sci. USA 92, 3586–3590.
11. R. Sandbrink, C. L. Masters, and K. Beyreuther (1994) J. Biol. Chem. 269, 1510–1517.
12. J. Kang et al. (1987) Nature 325, 733–736.
13. F. S. Esch et al. (1990) Science 248, 1122–1124.
14. P. Seubert et al. (1993) Nature 361, 260–263.
15. P. E. Fraser, J. Nguyen, W. Surewicz, and D. A. Kirschner (1991) Biophys. J. 60, 1190–1201.
16. D. A. Kirschner, C. Abraham, and D. A. Selkoe (1986) Proc. Natl. Acad. Sci. USA 83, 503507- .
17. P. E. Fraser et al. (1992) Biochemistry 31, 10716–10723.
18. H. Inouye, P. E. Fraser, and D. A. Kirschner (1993) Biophys. J. 64, 502–519.
19. M. Shoji et al. (1992) Science 258, 126–129.
20. C. Soto, E. Castano, B. Frangione, and N. Inestrosa (1995) J. Biol. Chem. 270, 3063–3067.
21. H. Sticht, P. Bayer, D. Willbold, S. Dames, C. Hilbich, K. Beyreuther, R. W. Frank, and P. Rosch (1995) Eur. J. Biochem. 233, 293–298.
22. R. Sherrington et al. (1995) Nature 375, 754–760.
23. E. Levy-Lahad et al. (1995) Science 269, 973–977.
24. E. Rogaev et al. (1995) Nature 376, 775–778.
25. M. Barinaga (1995) Science 268, 1845–1846.
26. E. H. Corder et al. (1993) Science 261, 921–923.

|
|
|
|
دراسة يابانية لتقليل مخاطر أمراض المواليد منخفضي الوزن
|
|
|
|
|
|
|
اكتشاف أكبر مرجان في العالم قبالة سواحل جزر سليمان
|
|
|
|
|
|
|
اتحاد كليات الطب الملكية البريطانية يشيد بالمستوى العلمي لطلبة جامعة العميد وبيئتها التعليمية
|
|
|
















