
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Cell Cycle |


|
|
Read More
Date: 22-11-2015
Date: 15-5-2016
Date: 18-6-2021
|
Cell Cycle
A dramatic change in how we understand the progression of the eukaryotic cell cycle, from phenomenological to biochemical, occurred in the late 1980s. The cell cycle consists of four main phases: G1, S, G2, and M. DNA replication occurs in S phase, and the chromosomes are segregated to daughter cells during mitosis (M phase). The so-called gap phases (G1 and G2) are defined simply as the periods separating DNA replication and mitosis. The length of the cell cycle ranges from just a few minutes in certain early embryos (which don't need to increase their mass between divisions), to 1.5 h in budding yeast, to approximately one day in typical mammalian tissue culture cells. In mammalian cells, the durations of S phase (~7 h) and mitosis (~1 h) are relatively inflexible, whereas the lengths of the gap phases can vary greatly. The length of G1 is particularly variable and is sensitive to the levels of nutrients and growth factors. In general, once a cell progresses past the “Restriction Point” in G1, it becomes committed to complete the cell cycle. A convergence of research in yeast, marine invertebrates, and frogs led to a phenomenal growth in our mechanistic understanding of how all eukaryotic cells regulate these events and the transitions between them. The basic machinery controlling the cell cycle consists of a subfamily of protein kinases, termed cyclin-dependent kinases (Cdk's). These enzymes are in turn regulated by a number of mechanisms, including transcription, inhibitory and activating phosphorylations, binding to inhibitory proteins, and ubiquitin-mediated protein degradation. Checkpoints that prevent the execution of one cellular event before a prior step has been completed further modulate cell cycle progression.
1. Historical Threads: Yeast Genetics, MPF, and Cyclins
Genetic studies of the cell cycle in the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe illuminated important principles underlying the logic of cell cycle control and identified many of its key players. Screens were performed for conditional temperature-sensitive mutations, causing cells to arrest quickly with uniform morphologies, indicative of defects in executing individual cell cycle steps. Analysis of a large collection of such cdc (cell division cycle) mutants from S. cerevisiae placed them into series of dependent and independent pathways (1). For example, inactivation of CDC28 causes cells to arrest at “START” (equivalent to the restriction point of mammalian cells) and blocks pathways leading to formation of the bud, DNA replication, and duplication of the spindle pole body. Inactivation of genes acting after CDC28 can block one of these pathways, while the other two continue. A similar screen in the distantly related yeast Schizosaccharomyces pombe successfully identified the most proximal regulators of the G2 to mitosis transition (Fig. 1) (2). cdc2+ was found to be the key regulator of entry into mitosis and to encode a protein kinase. Inactivation of cdc2+ causes cells to arrest in G2, whereas its premature activation leads to early entry into mitosis. Genetic analysis indicated that the cdc25+ gene product functioned in a pathway leading to activation of Cdc2 and that the product of wee1+ functioned in a pathway leading to inhibition of Cdc2. Sequence analysis of these genes indicated that both cdc2+ and wee1+ encoded protein kinases, but initially shed little light on the function of Cdc25.
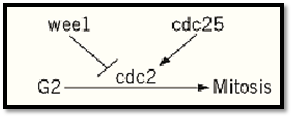
Figure 1. Regulation of the G2-to-mitosis transition in Schizosaccharomyces pombe. cdc2+ is required for this transition. In the absence of its activity, cells arrest in G2. cdc25+ is a positive regulator of cdc2+, and its inactivation also gives a G2 arrest. In contrast, wee1+ negatively regulates cdc2+, and its inactivation leads to premature activation of Cdc2 and entry of cells into mitosis at an unusually small size (a “wee” phenotype).
Two other lines of research provide essential historical threads leading to our current understanding of the cell cycle. The first involves maturation promoting factor (MPF), which was originally defined as a developmental factor found in the cytoplasm of metaphase-arrested frog eggs that could induce oocytes to proceed through meiosis and to “mature” into eggs following its injection into their cytoplasm (3). Subsequent research found that MPF activity is present in all eukaryotic cells during M phases (meiosis or mitosis). Thus, MPF is a universal regulator of entry into mitosis. The second thread concerns the cyclins. The first cyclins were found during studies of translational control before and after fertilization of sea urchin eggs conducted as part of the Physiology course at the Marine Biological Laboratory in Woods Hole (4). These proteins were synthesized continuously, and they accumulated until their abrupt degradation during mitosis. This pattern of accumulation hinted that cyclins might play an important role during the cell cycle. Later work showed that the injection of cyclin messenger RNA led to the maturation of frog oocytes, suggesting that cyclins, like MPF, functioned as positive regulators of the G2-to-mitosis transition (5).
Work on cyclins, MPF, and the yeast cdc genes came to an explosive convergence in the late 1980s with the purification of MPF and the identification of its subunits as homologs of cdc2+ and cyclin (6-9) .Thus, this key regulator of entry into mitosis was a protein kinase (Cdc2) whose activity was controlled at least in part via the cyclic accumulation and degradation of a regulatory subunit ) cyclin). The activity of Cdc2 is typically assayed by its ability to phosphorylate a convenient substrate, histone H1. Cdc2 is a workhorse master regulator. Rather than sitting at the apex of a large regulatory pathway, Cdc2 generally phosphorylates the downstream-most targets of mitotic regulation. For example, its direct phosphorylation of nuclear lamins causes the disassembly of the lamin-containing filaments of the nuclear lamina (10, 11).
2. Regulation of Cdc2 Activity: Phosphorylation and Cyclin Degradation
In addition to cyclin binding, enzymes such as Cdc2 are regulated by multiple phosphorylations (12,) (13see Fig. 2). Cdc2 is negatively regulated via phosphorylation of Thr14 and Tyr15. (The amino acid positions in this section refer to the phosphorylation sites in human Cdc2.) These sites are phosphorylated by Wee1 (the negative regulator of Cdc2 identified genetically in Schizosaccharomyces pombe (see Fig. 1)) and by Wee1-like protein kinases (Mik1 in Schizosaccharomyces pombe and the membrane-bound Myt1 protein in vertebrates). Dephosphorylation of these inhibitory sites is carried out by Cdc25 proteins, genetically identified activators of Cdc2 that share distant similarity to protein tyrosine phosphatases. Finally, activating phosphorylation of Cdc2 occurs on Thr161 within the so-called T-loop and is carried out by the Cdk-activating kinase (CAK). Dephosphorylation of Thr161 has not been as well studied. It occurs following cyclin degradation, possibly by a phosphatase termed KAP. Two feedback loops operate as cells enter mitosis and lead to the inhibition of Wee1-like enzymes and the stimulation of Cdc25, thus ensuring the abrupt and irreversible transition from G2 to mitosis. The roles of cyclin binding and activating phosphorylation have been studied using X-ray crystallography of Cdk2 (14-16), a close relative of Cdc2. Monomeric Cdk2 is inactive for two major reasons. First, residues that interact with the phosphates of ATP are out of position, resulting in the misalignment of the g-phosphate of ATP for phosphoryl transfer. Second, the T-loop is positioned so that it would physically prevent protein substrates from gaining access to the site of catalysis. Binding to cyclin relieves this steric block and moves residues that interact with ATP into their proper positions. The cyclin A-Cdk2 complex is about 1% active. Full activation requires activating phosphorylation (on Thr160 in Cdk2), which alters the positions of some T-loop residues and creates an acidic patch for the binding of optimal substrates containing a key basic residue.
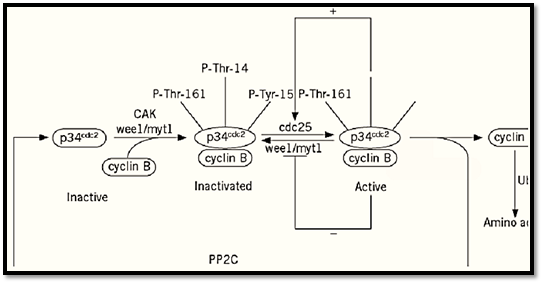
Figure 2. Activation of Cdc2. Monomeric Cdc2 is unphosphorylated and inactive. Binding to cyclin induces its phosphorylation on inhibitory sites (Thr14 and Tyr15) by Wee1-like protein kinases and its phosphorylation on an active site (Thr161) by CAK. The transition into mitosis is accompanied by two feedback loops involving stimulation of Cdc25 (the phosphatase acting on Thr14 and Tyr15) and inhibition of Wee1-like enzymes, resulting in the abrupt and irreversible activation of Cdc2 and entry into mitosis. The degradation of cyclin by the ubiquitin system toward the end of mitosis lea to the inactivation of Cdc2 and its dephosphorylation, probably by a phosphatase called KAP.
One of the irreversible ratchet steps in the cell cycle is the degradation of cyclins by the ubiquitin system (17). Ubiquitin is a 76-residue protein whose covalent attachment to proteins can target them for proteolysis by the proteasome, a huge multiproteinase unwinding and degrading machine. Ubiquitin is activated by its ATP-dependent covalent attachment to an enzyme called E1. E1 then transfers ubiquitin to one of many E2 enzymes. The E2s can ubiquitinate substrates, often with the help of an E3. The E3s may be the most diverse and interesting components of this system. Some E3s receive the covalently bound ubiquitin; others serve as matchmakers that bring together a substrate and the appropriate E2. For the mitotic cyclins, the E3 was first termed the cyclosome, though it is now generally termed the anaphase promoting complex (APC), an 8- to 12-subunit complex. The APC is the regulated component of the ubiquitin system for the degradation of the mitotic cyclins and serves as the target for checkpoint signals that can block cyclin degradation. The ubiquitin-dependent degradation of cyclins that act earlier in the cell cycle generally does not require the APC and has been best studied in S. cerevisiae.
3. Relatives: Cdks and Cyclins
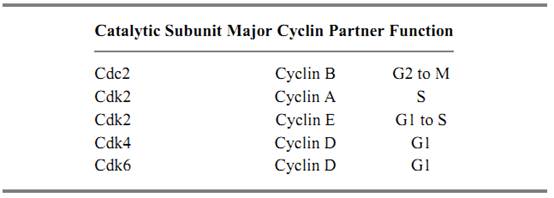
Both Cdc2 and the mitotic cyclins are members of large families of related proteins, many of which function in cell cycle regulation. The Cdc2 relatives are termed Cdks (for cyclin-dependent kinases( and the cyclin relatives are designated by letter. The different cyclins accumulate at various times during the cell cycle and pair with a limited subset of the Cdks to execute their respective functions. The major cyclin–Cdk complexes and their functions are shown in Table 1. Complexes of D-type cyclins with Cdk4 and Cdk6 function in progression through G1 and are the first kinases to phosphorylate the cell-cycle inhibitor retinoblastoma protein (Rb). The cyclin E–Cdk2 complex stimulates the initiation of DNA synthesis, and the cyclin A–Cdk2 complex is generally thought to promote passage through S phase. Cyclin B partners with Cdc2 to promote the G2-to-mitosis transition. All of these complexes are regulated by phosphorylations on sites equivalent to those used in Cdc2. In addition to these enzymes, a large number of cyclin–Cdk complexes function in processes other than cell cycle regulation, including transcription and neuronal differentiation. In general, the regulatory subunits of these Cdks are stable proteins and are classified as cyclins only on the basis of sequence similarity.
Table 1. Functions of Cyclin–Cdk Complexes Involved in Cell Cycle Control
4. Cdk Inhibitors
In addition to cyclin binding and phosphorylation, Cdks are also regulated by the binding of inhibitory proteins (18). These can be divided into two classes: the CIP/KIP family (consisting of p21CIP1, p27KIP1, and p27KIP2) and the INK4 family (consisting of p15INK4b, p16INK4a, p18INK4c, and p19INK4d). The CIP/KIP proteins bind preferentially to the cyclin–Cdk complex and cause its inactivation. These proteins bind to numerous Cdks, but show a preference for Cdk2. In contrast, the INK4 proteins bind preferentially to monomeric Cdks (inhibiting the binding of cyclin) and are specific for Cdk4 and Cdk6. Cdk inhibitors play very important roles both in cell cycle progression (particularly in responses to growth factors) and during development. One of the best-studied roles for a Cdk inhibitor involves the regulation of the initiation of DNA synthesis in S. cerevisiae by Sic1p. Entry into S phase in S. cerevisiae requires the activity of complexes containing an S-phase cyclin (Clb5p or Clb6p) and the budding yeast Cdk, Cdc28p. These complexes are inhibited by Sic1p. Degradation of Sic1p by the ubiquitin system is triggered following the phosphorylation of Sic1p by Cdc28p bound to G1 cyclins, thereby freeing the Clb5p and Clb6p complexes to initiation DNA replication (19).
5. Checkpoints
Among the key mechanisms for ensuring the orderly progression of cell cycle events are checkpoints, which monitor the completion of certain events and prevent the execution of subsequent events if the monitored event has not been completed (20, 21). For example, checkpoints can block entry into mitosis if the DNA is damaged or has not been fully replicated, or they can block exit from mitosis if the spindle has not been properly assembled. In S. cerevisiae a checkpoint even monitors whether a bud has formed. One of the defining features of a checkpoint sensor is that it is not part of the machinery being sensed and that it is dispensable, though only for the short term. The checkpoint that provides the paradigm for all others involves the sensing of DNA damage and unreplicated DNA by Rad9p in S. cerevisiae. A number of yeast cdc mutants cause G2 arrests that are dependent on this checkpoint. For example, inactivation of CDC9, which encodes DNA Ligase, leads to G2 arrest if RAD9 is functional. In contrast, inactivation of CDC9 in a rad9 mutant does not lead to an immediate arrest. Instead, cells progress into mitosis and continue through a couple of cell cycles, until they arrest as microcolonies. The G2-arrested RAD9 cells remain viable while they attempt to repair their DNA, whereas the rad9 cells rapidly lose viability as they proceed through the checkpoint. Unperturbed wild-type cells grow normally in the absence of this checkpoint. The importance of checkpoints to a proper cell cycle and to human health is underscored by the p53 protein. p53 is the most frequently mutated protein in human cancers and functions as part of a checkpoint responding to DNA damage by transcriptionally inducing the p21 Cdk inhibitor (22), thereby blocking cell cycle progression. The absence of this checkpoint leads to genomic instability ) the accumulation of further mutations, loss of chromosomes, etc.) and a greatly increased risk of tumor formation.
References
1. J. R. Pringle and L. H. Hartwell (1981) In The Molecular Biology of the Yeast Saccharomyces ) J. Strathern, E. Jones, and J. Broach, eds.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 97–142.
2. P. Nurse (1990) Nature 344, 503–508.
3. Y. Masui and C. L. Markert (1971) J. Exp. Zool. 177, 129–146.
4. T. Evans, E. T. Rosenthal, J. Youngblom, D. Distel, and T. Hunt (1983) Cell 33, 389–396.
5. K. I. Swenson, K. M. Farrell, and J. V. Ruderman (1986) Cell 47, 861–870.
6. W. G. Dunphy, L. Brizuela, D. Beach, and J. Newport (1988) Cell 54, 423–431.
7. J. Gautier, C. Norbury, M. Lohka, P. Nurse, and J. Maller (1988) Cell 54, 433–439.
8. J. C. Labbé, J. P. Capony, D. Caput, J. C. Cavadore, J. Derancourt, M. Kaghad, J. M. Lelias, A. Picard, and M. Dorée (1989) EMBO J. 8, 3053–3058.
9. J. Gautier, J. Minshull, M. Lohka, M. Glotzer, T. Hunt, and J. L. Maller (1990) Cell 60, 487–494 .
10. M. Peter, J. Nakagawa, M. Dorée, J. C. Labbé, and E. A. Nigg (1990) Cell 61, 591–602.
11. G. E. Ward and M. W. Kirschner (1990) Cell 61, 561–577.
12. M. J. Solomon (1993) Curr. Opin. Cell Biol. 5, 180–186.
13. M. J. Solomon and P. Kaldis (1998) In Results and Problems in Cell Differentiation, Vol. 22: Cell Cycle Control (M. Pagano, ed.), Springer, Heidelberg, pp. 79–109.
14. J. L. DeBondt, J. Rosenblatt, J. Jancarik, H. D. Jones, D. O. Morgan, and S.-H. Kim (1993) Nature 363, 595–602.
15. P. D. Jeffrey, A. A. Russo, K. Polyak, E. Gibbs, J. Hurwitz, J. Massagué, and N. P. Pavletich (1995) Nature 376, 313–320.
16. A. A. Russo, P. D. Jeffrey, and N. P. Pavletich (1996) Nat. Struct. Biol. 3, 696–700.
17. R. W. King, R. J. Deshaies, J.-M. Peters, and M. W. Kirschner (1996) Science 274, 1652–1659.
18. T. J. Soos, M. Park, H. Kiyokawa, and A. Koff (1998) In Results and Problems in Cell Differentiation, Vol. 22: Cell Cycle Control (M. Pagano, ed.), Springer, Heidelberg, pp. 111–131 .
19. R. M. Feldman, C. C. Correll, K. B. Kaplan, and R. J. Deshaies (1997) Cell 91, 221–230.
20. L. H. Hartwell and T. A. Weinert (1989) Science 246, 629–634.
21. S. J. Elledge (1996) Science 274, 1664–1672.
22. J. W. Harper, G. R. Adami, N. Wei, K. Keyomarsi, and S. J. Elledge (1993) Cell 75, 805–816.

|
|
|
|
"عادة ليلية" قد تكون المفتاح للوقاية من الخرف
|
|
|
|
|
|
|
ممتص الصدمات: طريقة عمله وأهميته وأبرز علامات تلفه
|
|
|
|
|
|
|
المجمع العلمي للقرآن الكريم يقيم جلسة حوارية لطلبة جامعة الكوفة
|
|
|
















