علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

Photochemistry : Carbonyl Compounds
 المؤلف:
William Reusch
المؤلف:
William Reusch
 المصدر:
Virtual Textbook of Organic Chemistry
المصدر:
Virtual Textbook of Organic Chemistry
 الجزء والصفحة:
............
الجزء والصفحة:
............
 2-9-2018
2-9-2018
 4183
4183




+
-
20
Photochemistry : Carbonyl Compounds
Conjugated ketones are often good sensitizers, thanks to the efficiency of intersystem crossing from the
n → π* singlet to the triplet. However, before discussing this application it will be helpful to consider the bonding structure of the carbonyl group itself. In the case of the simple compound formaldehyde, the Lewis formula consists of two C–H sigma bonds, a C–O sigma bond, a C=O pi bond and two non-bonding electron pairs.
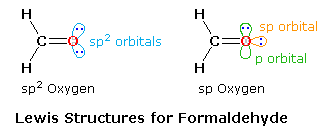
As shown in the diagram on the right, two Lewis structures, differing in the hybridization of oxygen, may be drawn, The structure on the left is a common representation in which an sp2 oxygen replaces one of the CH2 groups of an ethylene molecule. However, based on the principle that stable molecules will adopt the strongest bonding possible, the right hand structure becomes an attractive alternative.
An sp2–sp sigma bond should be stronger and shorter than a sp2–sp2 sigma bond, and the shorter bond distance will enhance the pi-bonding. Double bonds are of course shorter and stronger than equivalent single bonds. The average values given in the following table indicate that C=C is 13% shorter and 76% stronger than a C–C bond, whereas C=O is 15% shorter and over 100% stronger than a C–O bond, possibly reflecting the sp hybridization of the oxygen. An important difference between these models is found in the nature of the non-bonding electron pairs on oxygen. In the sp2-oxygen model these occupy very similar (degenerate) orbitals, but in the sp-oxygen model one pair is in a relatively low energy sp-orbital and the other in a higher energy p-orbital. Molecular orbital calculations clearly show that the latter model is a better representation than the former.
| Bond Length, Å | C–C, 1.54 | C=C, 1.34 | C–O, 1.44 | C=O, 1.22 |
| Bond Energy, kcal/mole | C–C, 84 | C=C, 146 | C–O, 86 | C=O, 177 |
A general diagram illustrating the two major electronic excitations of a carbonyl group is shown below. This terminology has been defined in the UV-Visible Spectroscopy chapter. The weak nature of the n → π* absorption is due to the poor overlap of the p-orbital housing the higher energy electron pair with the π* orbital (they are essentially orthogonal). In contrast, π and π* orbitals overlap almost completely, so the probability of a π → π* excitation being achieved by a photon of proper energy is high. This spatial overlap requirement has been termed a selection rule by spectroscopists. A second important selection rule concerns the probability of electronic transitions between states of different spin multiplicity, the spin selection rule. This rule reflects the high probability of transitions between different singlet states, or between different triplet states, but the low probability of singlet-triplet or triplet-singlet transitions. A table summarizing these rules is given below.
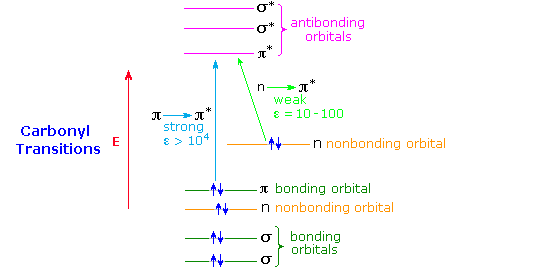
Formaldehyde, acetaldehyde and acetone show strong π → π* absorption at 180 to 190nm (ε = 103 to 104), and weak n → π* absorption at 280 to 300 nm (ε = 12 to 20). Conjugation with the π-electrons of a double bond or a benzene ring shifts both absorptions to longer wavelength and increases the strength of absorption.
| Compound | Cyclohexanone | (CH3)2C=CHCOCH3 | C6H5CHO | C6H5COCH3 |
|---|---|---|---|---|
| λmax π → π* | 200 nm (ε 2000) | 230 nm (ε 12,300) | 242 nm (ε 14,000) | 238 nm (ε 13,000) |
| λmax n→ π* | 285 nm (ε 14) | 325 nm (ε 90) | 328 nm (ε 50) | 320 nm (ε 40) |
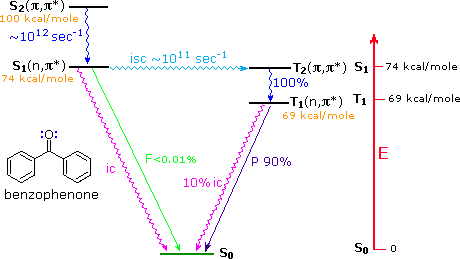
Aryl ketones such as acetophenone (right hand compound in the table above), undergo rapid intersystem crossing of the n → π* singlet excited state to an energetically close π → π* triplet state. The latter then quickly decays to the lower energy n → π* triplet, as shown for benzophenone in the diagram below. This circuitous route for the low probability direct conversion reflects modified selection rules formulated by Prof. M. A. El Sayed of UCLA (last two entries in the following table). This pathway is not available to most aliphatic ketones, so their intersystem crossing rates from n → π* singlets to triplets are slow.
As noted in the diagram, there is very little fluorescent decay from the S1 state, and radiationless decay to S0 is less than 1% of the intersystem crossing to T2. In the absence of quenching reactions, T1 returns to S0 by a mixture of phosphorescent and radiationless decay.
Benzophenone Excited States
| Transition | Probability | Factor |
|---|---|---|
| S0 → S1(n , π*) S0 → S1(π , π*) |
forbidden ---------------- allowed |
orbital overlap in space (also symmetry) |
| Sn → S1 → S0 Tn → T1 |
allowed | no change in spin multiplicity |
| S(n , π*) → T(n , π*) S(π , π*) → T(π , π*) |
forbidden | change in spin multiplicity |
| S(n , π*) → T(π , π*) S(π , π*) → T(n , π*) |
allowed | change in orbital configuration |
 الاكثر قراءة في كيمياء عامة
الاكثر قراءة في كيمياء عامة
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)