
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Alpha-Helix |


|
|
Read More
Date: 12-5-2021
Date: 30-3-2021
Date: 10-3-2021
|
Alpha-Helix (310-Helix and Pi-Helix)
The right-handed a-helix is one of two regular types of protein secondary structure, the other being the b-strand that forms b-sheets. Helices are formed from consecutive stretches of residues of the polypeptide chain and are characterized by a right-handed coiled backbone and a regular repeating pattern of backbone hydrogen bonds. a-Helices are usually depicted as coils or cylinders in protein structure diagrams (Fig. 1). There are 3.6 residues in every turn of a-helix, and for each turn the backbone is translated by 5.4 Å (or 1.5 Å per residue). In protein structures, the average length of an a-helix is 10 residues, although much shorter and longer examples have been observed. The backbone angles are approximately –60° and –50° for f and Y, respectively, corresponding to the allowed region in the lower left of the Ramachandran Plot. The atoms of the polypeptide chain pack closely together in the a-helical conformation, making favorable van der Waals interactions. The side chains of each residue are oriented outward from the axis of the helix, with the Ca–Cb bond pointing toward the N-terminal end of the helix.
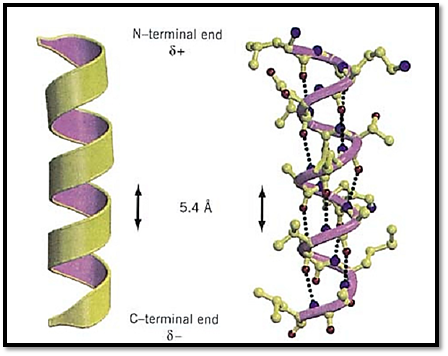
Figure 1. A typical protein a-helix. (Left) The helix is depicted schematically as a coil, and the N- and C-terminal ends, with their respective partial positive and negative charges, are indicated. (Right) The atomic detail of the a-helical structure is shown, with hydrogen bonds between backbone carbonyl oxygens (i) and backbone amide nitrogens of residue (i + 4) indicated by dotted lines. Nitrogen atoms are shown in blue and oxygen atoms are shown in red. This figure was generated using Molscript (3) and Raster3D (4, 5). See color insert.
A regular hydrogen bond pattern is formed in the a-helix, corresponding to bonds formed between the backbone carbonyl oxygen of residue (i) and the backbone amide of residue (i + 4) in the polypeptide chain, so that there are 13 atoms between each acceptor and donor pair. Thus, apart from the first few amide groups and last few carbonyl oxygen atoms, all the peptide bonds in an a-helix form hydrogen bond interactions. The hydrogen bonds are all oriented in the same direction, as are the dipoles of each of the peptide bonds. Therefore, the helix itself also has a significant dipole moment (a partial positive charge at the N-terminus and a partial negative charge at the C-terminus), and charged residues that interact with the dipole are often found in the sequence at the appropriate end of a helix (1). For example, the negatively charged Asp residue is highly favored at the N-terminus. The helix dipole has also been implicated in protein functions, such as substrate binding and catalysis.
Different amino acids have different tendencies for forming a-helices (2). The amide nitrogen of proline is cyclized with its backbone and thus cannot act as a hydrogen bond donor. Proline residues therefore are not found frequently in a-helices; when they are, they cause irregularities such as kinks and bends in the middle of helices. On the other hand, proline is a preferred residue at the N-terminus of the helix where other residues would have an unpaired backbone amide.
The staggered arrangement of side chains around the helix can be visualized in two dimensions through the use of a helical wheel (a projection down the axis of the helix). Helices in membrane proteins are often amphipathic, having polar or charged side chains arranged on one side and hydrophobic side chains on the opposite side of the helix.
There are several variations to the a-helix conformation of proteins. The 310-helix is more tightly wound than the a-helix with backbone hydrogen bonds formed between residues (i) and (i + 3). Its name is based on its structure, with three residues per turn and 10 atoms between hydrogen bond donor and acceptor (although in real protein structures this geometry may be somewhat distorted). The 310-helix is usually only observed at the ends of a-helices or in short stretches of 4 to 5 residues. The P-helix is more loosely wound than the a-helix with backbone hydrogen bonds formed between the (i) and (i + 5) residues and is very rarely observed in protein structures. The left-handed helix, having the same hydrogen bonding pattern as a-helices but backbone f and Y angles of +60° and +50° (rather than the –60° and –50° angles of the right-handed a-helix), is not often observed, because this conformation results in steric clashes between backbone and side chain atoms. A glycine residue, however, having only a hydrogen atom as its side chain, can adopt this conformation. The poly(Pro)II helix, with no backbone hydrogen bonds, is also left-handed, with three residues per turn and a translation of 3.12 Å per residue. This helical structure is observed in polyproline and in proline-rich regions of proteins.
References
1. L. Serrano and A. R. Fersht (1989) Nature 342, 296–299.
2. J. S. Richardson and D. C. Richardson (1988) Science 240, 1648–1652.
3. P. J. Kraulis (1991) J. Appl. Crystallogr. 24, 946–950.
4. E. A. Merritt and M. E. P. Murphy (1994) Acta Crystallogr. D50, 869–873.
5. D. J. Bacon and W. F. Anderson (1988) J. Mol. Graphics 6, 219–222.

|
|
|
|
"عادة ليلية" قد تكون المفتاح للوقاية من الخرف
|
|
|
|
|
|
|
ممتص الصدمات: طريقة عمله وأهميته وأبرز علامات تلفه
|
|
|
|
|
|
|
المجمع العلمي للقرآن الكريم يقيم جلسة حوارية لطلبة جامعة الكوفة
|
|
|
















