آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Phenylketonuria |


|
|
Read More
Date: 12-11-2021
Date: 13-10-2021
Date: 26-11-2021
|
Phenylketonuria
PKU is the most common clinically encountered inborn error of amino acid metabolism (incidence 1:15,000). It is caused by a deficiency of PAH (Fig. 1). Biochemically, PKU is characterized by hyperphenylalaninemia.
Phenylalanine is present in high concentrations (ten times normal) not only in plasma but also in urine and body tissues. Tyrosine, which normally is formed from phenylalanine by PAH, is deficient. Treatment includes dietary restriction of phenylalanine and supplementation with tyrosine. [Note: Hyperphenylalaninemia may also be caused by rare deficiencies in any of the several enzymes required to synthesize BH4 or in dihydropteridine reductase, which regenerates BH4 from BH2 (Fig. 2).
Such deficiencies indirectly raise phenylalanine concentrations, because PAH requires BH4 as a coenzyme. BH4 is also required for tyrosine hydroxylase and tryptophan hydroxylase, which catalyze reactions leading to the synthesis of neurotransmitters, such as serotonin and the catecholamines. Simply restricting dietary phenylalanine does not reverse the central nervous system effects due to deficiencies in neurotransmitters.
Supplementation with BH4 and replacement therapy with L-3,4-dihydroxyphenylalanine and 5-hydroxytryptophan (products of the affected tyrosine hydroxylase– and tryptophan hydroxylase–catalyzed reactions) improves the clinical outcome in these variant forms of hyperphenylalaninemia, although the response is unpredictable.]
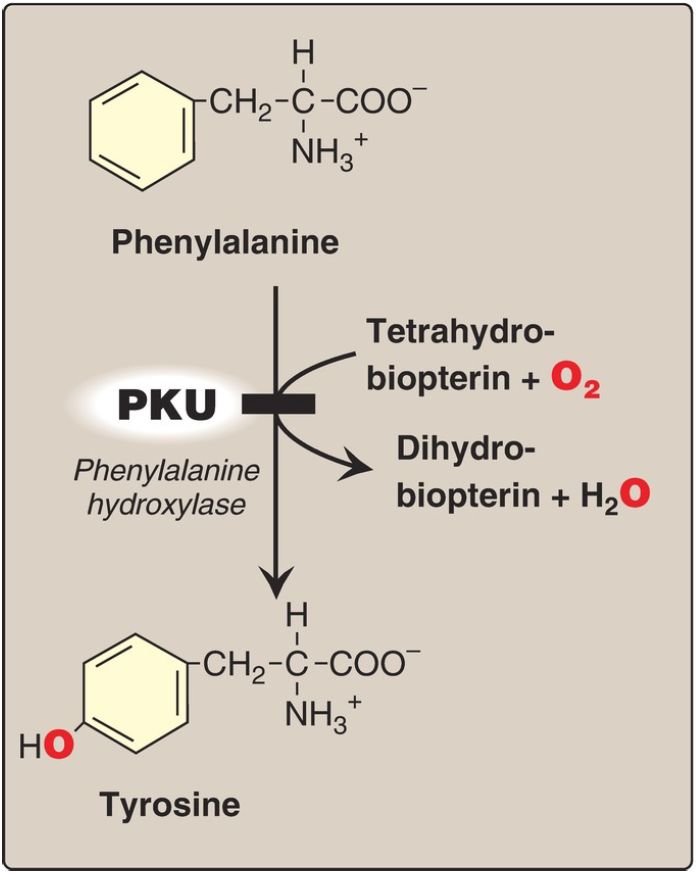
Figure 1: A deficiency in phenylalanine hydroxylase results in the disease phenylketonuria (PKU).

Figure 2: Biosynthetic reactions involving amino acids and tetrahydrobiopterin. [Note: Aromatic amino acid hydroxylases use BH4 and not PLP (pyridoxal phosphate).] NAD(H) = nicotinamide adenine dinucleotide; GTP = guanosine triphosphate; DOPA = L-3,4-dihydroxyphenylalanine; O2 = oxygen.
Screening of newborns for a number of treatable disorders, including inborn errors of amino acid metabolism, is done by tandem mass spectrometry of blood obtained from a heel prick. By law, all states must screen for >20 disorders, with some screening for >50. All states screen for PKU.
1. Additional characteristics: As the name suggests, PKU is also characterized by elevated levels of a phenylketone in the urine.
a. Elevated phenylalanine metabolites: Phenylpyruvate (a phenylketone), phenylacetate, and phenyllactate, which are not normally produced in significant amounts in the presence of functional PAH, are elevated in PKU (Fig. 3). These metabolites give urine a characteristic musty (“mousy”) odor.
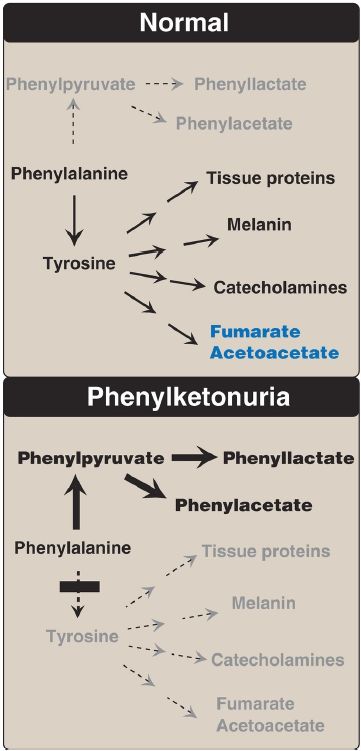
Figure 3: Pathways of phenylalanine metabolism in normal individuals and in patients with phenylketonuria.
b. Central nervous system effects: Severe intellectual disability, developmental delay, microcephaly, and seizures are characteristic findings in untreated PKU. The affected individual typically shows symptoms of intellectual disability by age 1 year and rarely achieves an intelligence quotient (IQ) >50 (Fig. 4). [Note: These clinical manifestations are now rarely seen as a result of newborn screening programs, which allow early diagnosis and treatment.]

Figure 4: Typical intellectual ability in untreated patients of different ages with phenylketonuria. IQ = intelligence quotient.
c. Hypopigmentation: Patients with untreated PKU may show a deficiency of pigmentation (fair hair, light skin color, and blue eyes).
The hydroxylation of tyrosine by copper-requiring tyrosinase, which is the first step in the formation of the pigment melanin, is decreased in PKU because tyrosine is decreased.
2. Newborn screening and diagnosis: Early diagnosis of PKU is important because the disease is treatable by dietary means. Because of the lack of neonatal symptoms, laboratory testing for elevated blood levels of phenylalanine is mandatory for detection. However, the infant with PKU frequently has normal blood levels of phenylalanine at birth because the mother clears increased blood phenylalanine in her affected fetus through the placenta. Normal levels of phenylalanine may persist until the newborn is exposed to 24–48 hours of protein feeding. Thus, screening tests are typically done after this time to avoid false negatives. For newborns with a positive screening test, diagnosis is confirmed through quantitative determination of phenylalanine levels.
3. Prenatal diagnosis: Classic PKU is caused by any of 100 or more different mutations in the gene that encodes PAH. The frequency of any given mutation varies among populations, and the disease is often doubly heterozygous (that is, the PAH gene has a different mutation in each allele). Despite this complexity, prenatal diagnosis is possible .
4. Treatment: Because most natural protein contains phenylalanine, an essential amino acid, it is impossible to satisfy the body’s protein requirement without exceeding the phenylalanine limit when ingesting a normal diet. Therefore, in PKU, blood phenylalanine level is maintained close to the normal range by feeding synthetic amino acid preparations free of phenylalanine, supplemented with some natural foods (such as fruits, vegetables, and certain cereals) selected for their low phenylalanine content. The amount is adjusted according to the tolerance of the individual as measured by blood phenylalanine levels. The earlier treatment is started, the more completely neurologic damage can be prevented. Individuals who are appropriately treated can have normal intelligence. [Note: Treatment must begin during the first 7–10 days of life to prevent cognitive impairment.] Because phenylalanine is an essential amino acid, overzealous treatment that results in blood phenylalanine levels below normal is avoided. In patients with PKU, tyrosine cannot be synthesized from phenylalanine, and, therefore, it becomes an essential amino acid and must be supplied in the diet.
Discontinuance of the phenylalanine-restricted diet in early childhood is associated with poor performance on IQ tests. Adult PKU patients show deterioration of IQ scores after discontinuation of the diet (Fig. 5). Therefore, lifelong restriction of dietary phenylalanine is recommended. [Note: Individuals with PKU are advised to avoid aspartame, an artificial sweetener that contains phenylalanine.]
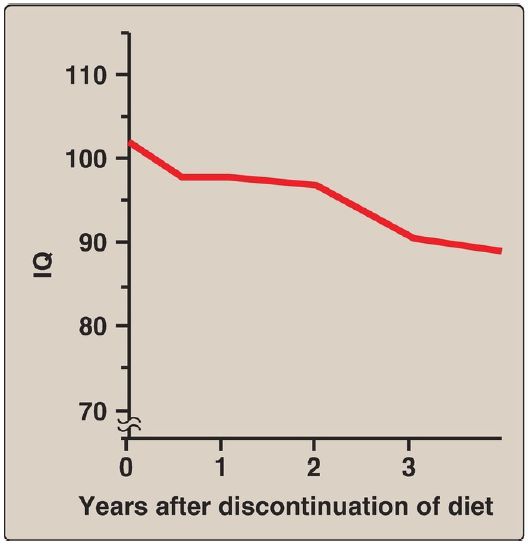
Figure 5: Changes in intelligence quotient (IQ) scores after discontinuation of low-phenylalanine diet in patients with phenylketonuria.
5. Maternal phenylketonuria: If women with PKU who are not on a lowphenylalanine diet become pregnant, the offspring can be affected with maternal PKU syndrome. High blood phenylalanine in the mother has a teratogenic effect, causing microcephaly and congenital heart abnormalities in the fetus. Because these developmental responses to high phenylalanine occur during the first months of pregnancy, dietary control of blood phenylalanine must begin prior to conception and be maintained throughout the pregnancy.

|
|
|
|
مخاطر خفية لمكون شائع في مشروبات الطاقة والمكملات الغذائية
|
|
|
|
|
|
|
"آبل" تشغّل نظامها الجديد للذكاء الاصطناعي على أجهزتها
|
|
|
|
|
|
|
تستخدم لأول مرة... مستشفى الإمام زين العابدين (ع) التابع للعتبة الحسينية يعتمد تقنيات حديثة في تثبيت الكسور المعقدة
|
|
|