آخر المواضيع المضافة

 علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية
علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية | A closer examination of Huckel’s Rule and the Polygon Rule |


|
|
Read More
Date: 8-9-2019
Date: 14-11-2019
Date: 1-8-2018
|
More than 100 years ago, Kekule recognized the possible existence of other conjugated cyclic polyalkenes, which at least superficially would be expected to have properties like benzene. The most interesting of these are cyclobutadiene, 23 , and cyclooctatetraene, 24:
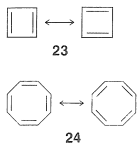
For each we can write two equivalent planar VB structures, and the qualitative VB method would suggest that both compounds, like benzene, have substantial electron-delocalization energies. However, the planar structures would have abnormal C−C=C
angles, and consequently at least some degree of destabilization associated with these bond angles. Nonetheless, estimation of the strain energies show that while they are substantial, they are not prohibitive. Should then these molecules be stabilized by resonance in the same sense as benzene is postulated to be?
In 1911 a German chemist, R. Willstatter (Nobel Prize 1915), reported an extraordinary thirteen-step synthesis of cyclooctatetraene from a rare alkaloid called pseudopelletierine isolated from the bark of pomegranate trees. The product was reported to be a light-yellow, highly unsaturated compound that absorbed four moles of hydrogen to form cyclooctane. Numerous tries to repeat the Willstatter synthesis were unsuccessful, and in the 1930s the prevailing opinion was that the product had been misidentified. However, during the Second World War, the German chemist W. Reppe found that cyclooctatetraene can be made in reasonable yields by the tetramerization of ethyne under the influence of a nickel cyanide catalyst:

The properties of the product substantiated Willstatter’s reports and it became clear that cyclooctatetraene is not like benzene.
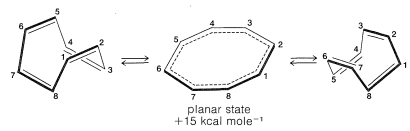
Subsequent studies of the geometry of the molecule revealed further that it is nonplanar, with alternating single and double bonds, 25a:
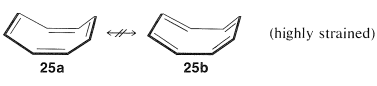
This geometry precludes the possibility of two equivalent VB structures, as for benzene, because, as you will see if you try to make a ball-and-stick model, 25b is highly strained and not energetically equivalent to 25a at all. Thus we can conclude that the delocalization energy of cyclooctatetraene is not large enough to overcome the angle strain that would develop if the molecule were to become planar and allow the π electrons to form equivalent π bonds between all of the pairs of adjacent carbons.
Cyclobutadiene, 23 , eluded Kekule, Willstatter, and a host of other investigators for almost 100 years. As more work was done, it became increasingly clear that the molecule, when formed in reactions, was immediately converted to something else. Finally, the will-o’-the-wisp was captured in an essentially rigid matrix of argon at 8K. It was characterized by its spectral properties (not by combustion analysis). On warming to even 35K, it dimerizes to yield 26 :
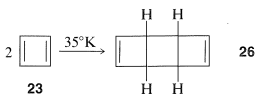
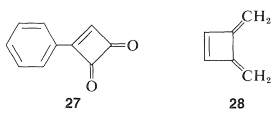
One possibility for the lack of stability4 of cyclobutadiene is that the angle strain associated with having four sp2 carbons in a four-membered ring is much greater than estimated. However, the stable existence of many compounds with four such sp2 carbons, for example 27 and 28 , make this argument weak, if not invalid:
Why, then, is cyclobutadiene so unstable and reactive? On this point, and also with respect to the nonaromatic character of cyclooctatetraene, the simple qualitative VB method that we have outlined is no help whatsoever. There is no way simply to look at the electron-pairing schemes 23 and 24 and see any difference between them and the corresponding schemes for benzene.
It is in this area that qualitative MO procedures have great success because there are general characteristics of the π molecular orbitals of monocyclic, conjugated polyene systems that predict differences in the properties of cyclobutadiene, benzene, cyclooctatetraene, and other similar compounds that are not obvious from the simple VB method.
As a rule, for N parallel atomic p orbitals overlapping in the π manner in a monocyclic array, there will be just one lowest molecular orbital, with all the atomic orbitals having the same phase. This will be seen for benzene in Figure 21-5. What is harder to understand without going through the calculations is that the higher-energy molecular orbitals for cyclic conjugated polyenes are predicted to come in successive degenerate6 pairs, as shown in Figure 21-13 for N=3 to 9.
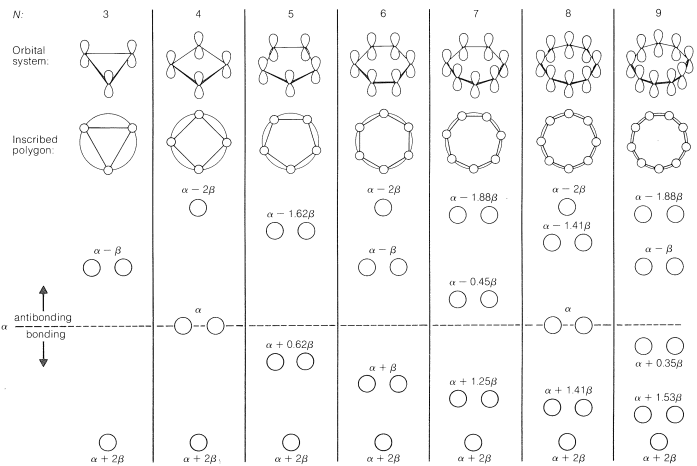
Figure 1: Calculated molecular-orbital energies of planar cyclic π -orbital systems made up of N 2p carbon atomic orbitals, in units of α and β
The qualitative ordering and, indeed, the numerical values of the energies of the π molecular orbitals for a cyclic system of N p orbitals can be derived in a very simple way. It is necessary only to inscribe a regular polygon with N sides inside a circle of radius 2β with a corner down. For example, for N=5 we get the following:
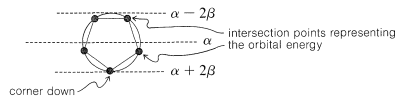
The molecular orbital energies are in units of β at the corners of the polygon. The nonbonding level corresponds to the horizontal dashed line drawn through the center of the circle.
The data of Figure 1. provide a rationale for the instability of cyclobutadiene and cyclooctatetraene. For cyclobutadiene, we can calculate that four π electrons in the lowest orbitals will lead to a predicted π-electron energy of 2(α+2β)+2(α)=4α+4β, which is just the π-electron energy calculated for two ethene bonds. The delocalization energy of the π
electrons of cyclobutadiene therefore is predicted to be zero!
Another feature of the π system of cyclobutadiene is that the four π electrons do not suffice to fill the three lowest orbitals and, if we apply Hund’s rule, the best way to arrange the electrons is as in 29, with two unpaired electrons, which is known as a triplet state:7
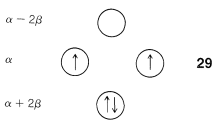
With the MO predictions of zero delocalization energy and an electronic configuration with unpaired electrons, we should not be surprised that cyclobutadiene readily dimerizes to give 26
even at very low temperatures.
The energies of the molecular orbitals calculated for planar cyclooctatetraene (Figure 1.) lead to a predicted delocalization energy of (8α+9.64β)−(8α+8β)=1.64β (∼31kcal), which is smaller than that of benzene, even though there are eight atomic orbitals instead of six through which the electrons are delocalized. Furthermore, the lowest electronic configuration for the planar molecule is, like cyclobutadiene, predicted to be a triplet. Experimental evidence indicates that the positions of the double bonds of cyclooctatetraene shift slowly as the result of formation of the molecule in the unstable planar state. The energy input required to flatten the molecule is about 15kcal mol−1
:
Because the bonding molecular orbitals for π systems such as in Figure 1. will be just filled with 2, 6, or 10 electrons to give singlet states, and 4 or 8 electrons would give triplet states, a (4n+2) π-electron rule was formulated for stable configurations and a 4n π-electron rule for unstable configurations, where n is an integer. Thus 2, 6, 10, 14, ⋯ π electrons will be favorable and 4, 8, 12 ⋯ π electrons will be unfavorable. This rule is the work of the German theoretician, E. Huckel, who devised the simple form of molecular orbital theory we have described in this chapter. The theory is appropriately called Huckel MO theory, and the rule is Huckel’s 4n+2 rule.
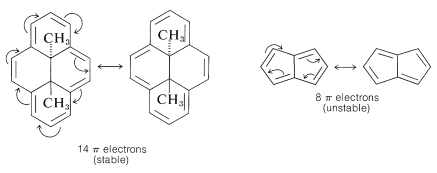
As Huckel formulated, the 4n+2n rule applies only to monocyclic systems. However, as a practical matter it can be used to predict the properties of polycyclic conjugated polyenes, provided the important VB structures involve only the perimeter double bonds, as in the following examples:
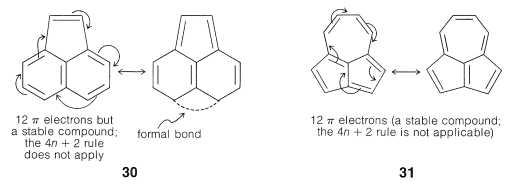
Application of the 4n+2 rule to other π systems, such as 30 and 31m, is not valid because good VB structures cannot be written that involve changes in the pairing schemes of the perimeter electrons all at once.


|
|
|
|
مخاطر خفية لمكون شائع في مشروبات الطاقة والمكملات الغذائية
|
|
|
|
|
|
|
"آبل" تشغّل نظامها الجديد للذكاء الاصطناعي على أجهزتها
|
|
|
|
|
|
|
تستخدم لأول مرة... مستشفى الإمام زين العابدين (ع) التابع للعتبة الحسينية يعتمد تقنيات حديثة في تثبيت الكسور المعقدة
|
|
|