آخر المواضيع المضافة

 علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية
علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية | Regioselectivity in Enolate Anion Formation and Reaction |


|
|
Read More
Date: 15-12-2016
Date: 22-9-2019
Date: 29-5-2017
|
Regioselectivity in Enolate Anion Formation and Reaction
The importance of enolate anions as synthetic intermediates is well established. Nevertheless, problems remain concerning their selective formation and reaction. For example, aldehyde enolate bases are likely to undergo the aldol reaction during their formation, and ketones like 2-heptanone have two different alpha-carbons, each capable of enolization. The ambident nature of enolate anions also enables electrophilic attack at both oxygen and carbon, but in most synthesis applications bonding to carbon is desired. Finally, enolate anions may often be formed as E/Z stereoisomers, and it has been shown that reaction stereoselectivity, when new chiral centers are created, depends on the enolate configuration.
The following diagram illustrates how the conditions under which enolate anion formation is accomplished can influence the regioselectivity of the reaction. The two ketone substrates, 2-heptanone and 2-methylcyclohexanone, each have differently substituted alpha-carbons. In each case, enolate anion mixtures are generated by reaction with a strong 2º-amide base (LDA is the usual choice). If the ketone is added to a cold THF solution of excess base, enolate anion formation is fast and irreversible (procedure a). On the other hand, if a slight excess of ketone is allowed to remain in solution, an equilibrium involving the ketone and the various enolate species is established (procedure b). At equilibrium the more stable enolate anion will predominate. The examples given in the diagram also report results from an equilibrating preparation in which the lithium metal in LDA is replaced by potassium (procedure c).
Regioselective Formation of Enolate Anions
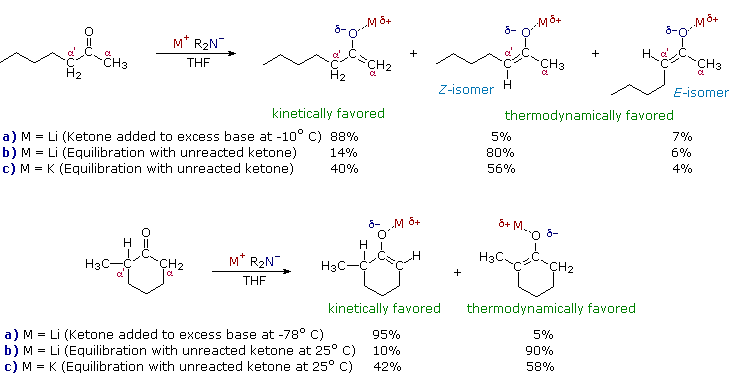
Several important principles are demonstrated here. First, if the enolate species has substantial double bond character, the more highly-substituted enolate double bond should predominate at equilibrium, as predicted from the stabilities of substituted alkenes. Since lithium-oxygen bonds are more covalent (have less ionic character) than potassium-oxygen bonds, the lithium enolate approximates an alkene more closely than the potassium enolate. Second, the greater ionic character of the potassium enolate places an increased negative charge on the alpha-carbon, a condition that is disfavored by alkyl group substitution. Indeed, the stability order of substituted carbanions is opposite to that of carbocations thanks to the electron donating character of alkyl groups relative to hydrogen. Finally, the rate of proton removal from an alpha-carbon site is decreased by alkyl substitution, probably reflecting a combination of steric hindrance (to bulky bases) and decreased carbanion stability. In both of the examples shown above, the conditions used in procedure (a) are typical of kinetically favored enolate formation, whereas those used in procedure (b) favorthermodynamic enolate formation. The comparative acidities provided by pKa values are derived from measurements made under equilibrating conditions, and therefore reflect thermodynamic acidity. Determinations of kinetic acidity require competitive isotope exchange experiments.
These principles influence the course of enolate alkylation reactions, as shown in the following diagram. In the first case, 2-methylcyclohexanone is converted to a thermodynamic enolate mixture, which is then reacted with methyl iodide. The major product is the expected 2,2-dimethylcyclohexanone (from the more stable enolate anion), but this is accompanied by di- and trimethylated products together with about 20% unreacted starting material. The complexity of the product mixture is due to acid-base proton transfer between alkylated products and unreacted enolate anion. In other words, once a small amount (say 5%) of dimethylcyclohexanone is formed, it finds itself in solution with a relatively high concentration of a strong base (the remaining enolate anion) that can remove another alpha-proton, giving a new enolate anion that is further methylated. If the kinetically favored lithium enolate (see the previous diagram) is used instead of the equilibrium potassium enolates, 2,6-dimethylcyclohexanone is the chief product.
The second reaction is an intramolecular alkylation that can occur in two different ways. If the kinetically favored enolate (methyl proton removal) is formed at low temperature, it reacts rapidly on warming to form a seven-membered ring. Alternatively, the weaker base, potassium tert-butoxide (in the alcohol as solvent), generates an equilibrium mixture of enolates which eventually react by intramolecular alkylation. The thermodynamically favored α'-enolate predominates, and the resulting alkylation generates a five-membered ring.


|
|
|
|
دخلت غرفة فنسيت ماذا تريد من داخلها.. خبير يفسر الحالة
|
|
|
|
|
|
|
ثورة طبية.. ابتكار أصغر جهاز لتنظيم ضربات القلب في العالم
|
|
|
|
|
|
|
قسم الشؤون الفكرية ينظم ندوة ثقافية بذكرى فاجعة هدم قبور أئمة البقيع (عليهم السلام)
|
|
|